
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
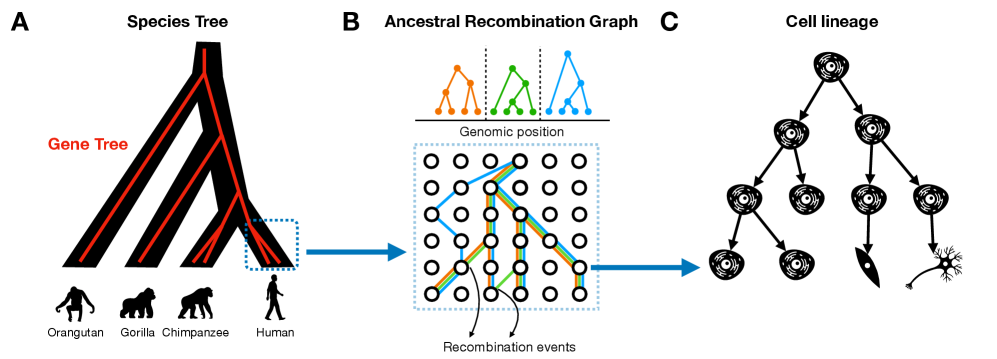
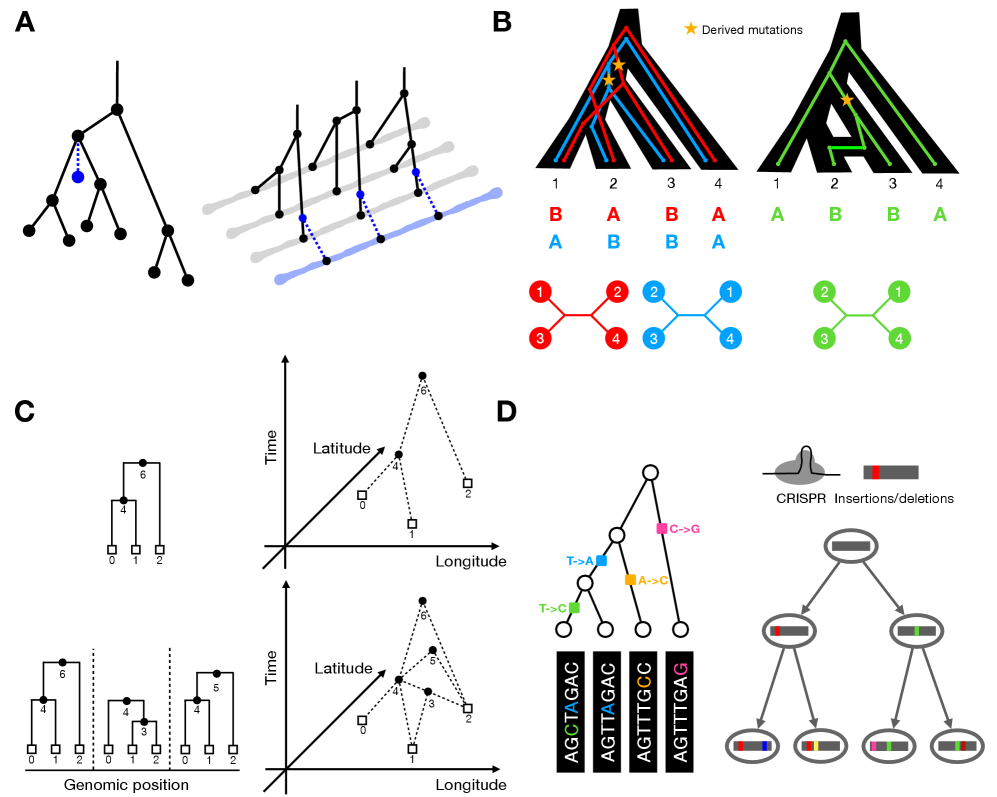
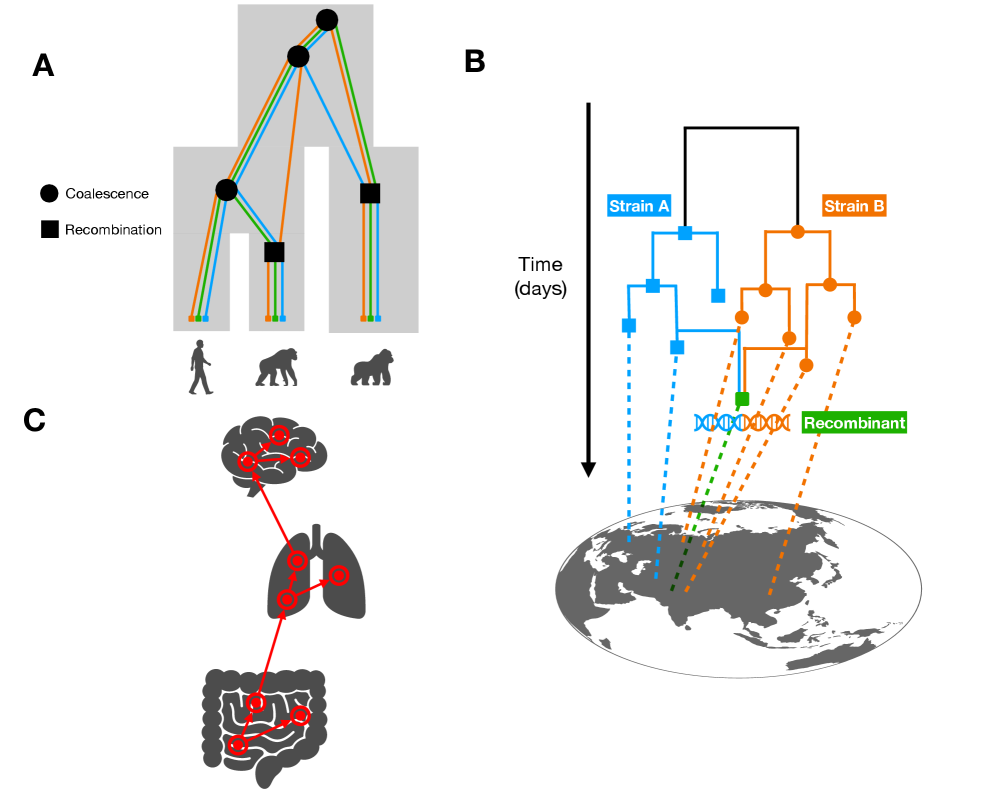
Tree Thinking in the Genomic Era: Unifying Models Across Cells, Populations, and Species
Stanford University | University of Oxford | University of California, Berkeley | Peking University | Guangzhou Medical University
30秒速读
IN SHORT: This paper addresses the fragmentation of tree-based inference methods across biological scales by identifying shared algorithmic principles and statistical challenges in phylogenetics, population genetics, and cell lineage tracing.
核心创新
- Methodology Identifies deep conceptual parallels between phylogenetic placement algorithms and ARG threading methods, demonstrating how phylogenetic placement generalizes to ARG reconstruction.
- Biology Shows that quartet-based network methods in phylogenetics and ABBA-BABA statistics in population genetics capture the same underlying signal of gene flow through asymmetric genealogical relationships.
- Methodology Demonstrates how ARG-based migration inference methods (e.g., GAIA, spacetrees) extend classical phylogeographic approaches by leveraging the full sequence of locally correlated genealogies along the genome.
主要结论
- Tree-based models provide a unified framework for ancestry inference across biological scales, with ARGs representing ~2.48 million SARS-CoV-2 genomes demonstrating pandemic-scale feasibility.
- Methodological parallels exist across domains: phylogenetic placement algorithms share core logic with ARG threading, and quartet-based methods in phylogenetics mirror ABBA-BABA statistics in population genetics for detecting gene flow.
- Current ARG inference algorithms remain constrained by simplifying assumptions (neutrality, panmixia, constant population size) and face challenges in uncertainty quantification, particularly for non-model species or limited sample sizes.
摘要: The ongoing explosion of genome sequence data is transforming how we reconstruct and understand the histories of biological systems. Across biological scales–from individual cells to populations and species–trees-based models provide a common framework for representing ancestry. Once limited to species phylogenetics, “tree thinking” now extends deeply to population genomics and cell biology, revealing the genealogical structure of genetic and phenotypic variation within and across organisms. Recently, there have been great methodological and computational advances on tree-based methods, including methods for inferring ancestral recombination graphs in populations, phylogenetic frameworks for comparative genomics, and lineage-tracing techniques in developmental and cancer biology. Despite differences in data types and biological contexts, these approaches share core statistical and algorithmic challenges: efficiently inferring branching histories from genomic information, integrating temporal and spatial signals, and connecting genealogical structures to evolutionary and functional processes. Recognizing these shared foundations opens opportunities for cross-fertilization between fields that are traditionally studied in isolation. By examining how tree-based methods are applied across cellular, population, and species scales, we identify the conceptual parallels that unite them and the distinct challenges that each domain presents. These comparisons offer new perspectives that can inform algorithmic innovations and lead to more powerful inference strategies across the full spectrum of biological systems.