
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
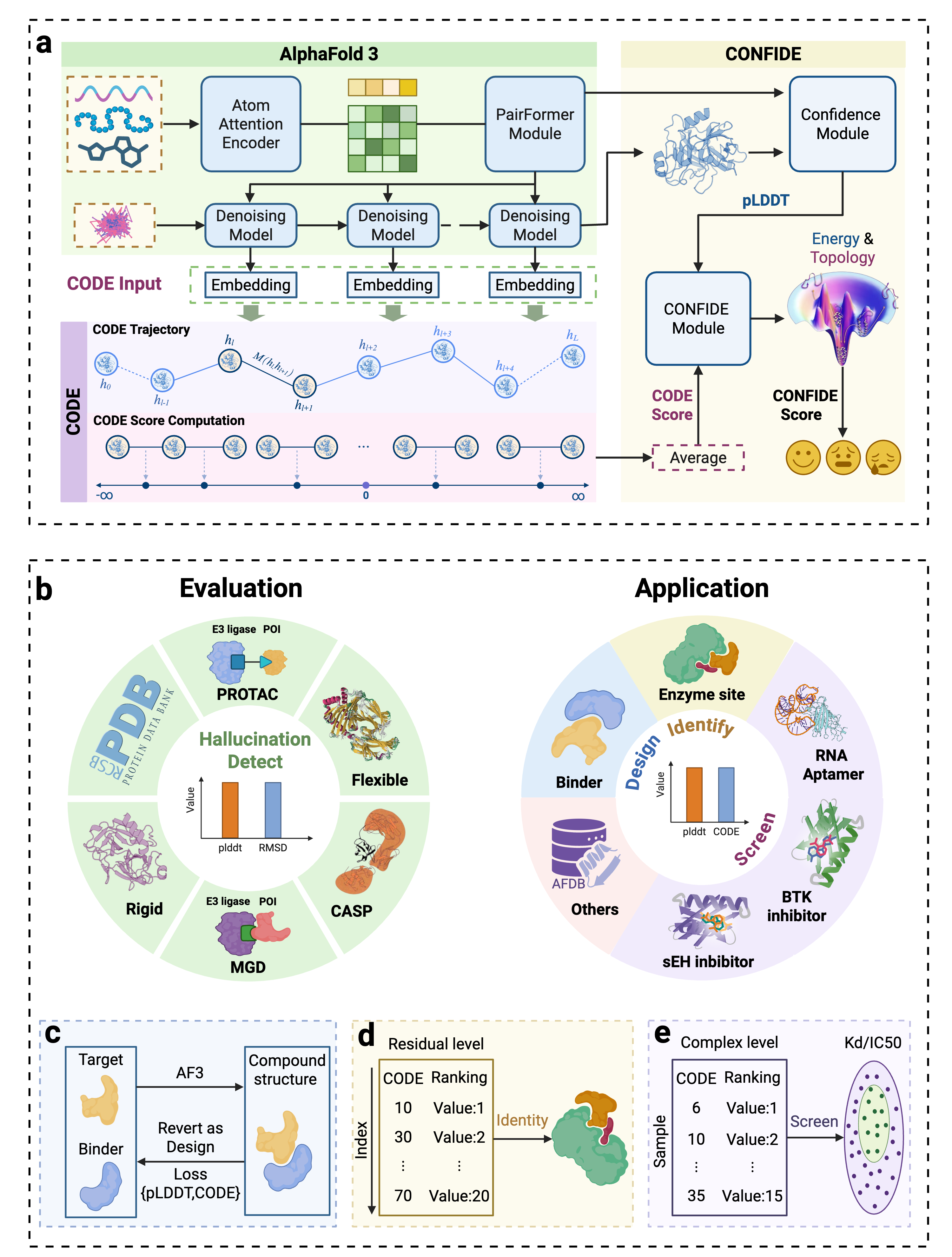
CONFIDE: Hallucination Assessment for Reliable Biomolecular Structure Prediction and Design
The Chinese University of Hong Kong | Zhejiang University | Macao Polytechnic University | University of Electronic Science and Technology of China
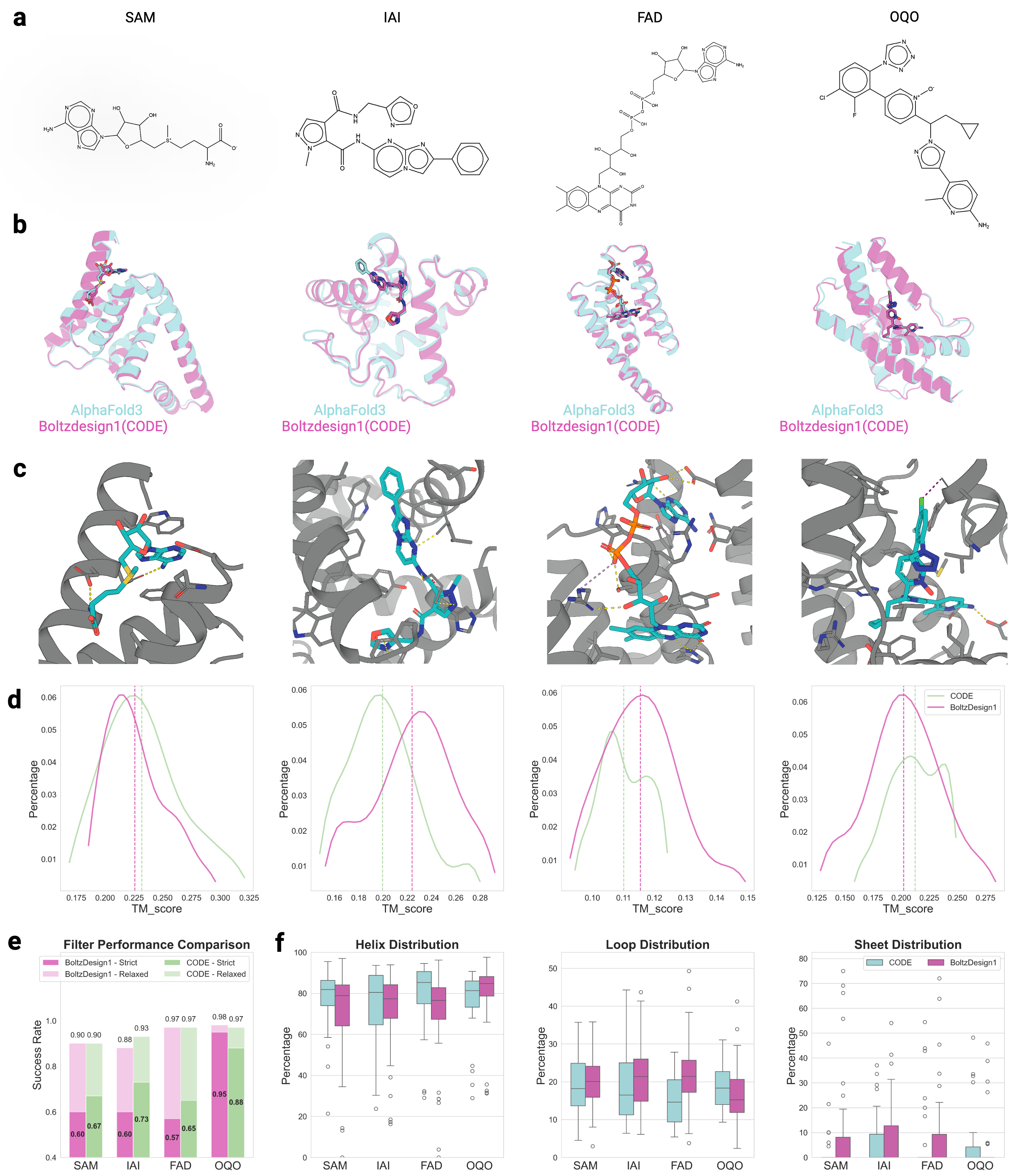
30秒速读
IN SHORT: This paper addresses the critical limitation of current protein structure prediction models (like AlphaFold3) where high-confidence scores (pLDDT) can be misleading, failing to detect subtle structural errors like atomic clashes and topological traps, which undermines reliability in downstream applications like drug discovery.
核心创新
- Methodology Introduces CODE (Chain of Diffusion Embeddings), a novel, unsupervised metric derived from AlphaFold3's latent diffusion embeddings that directly quantifies topological frustration, a key factor in protein folding kinetics previously overlooked by confidence scores.
- Methodology Proposes CONFIDE, a unified evaluation framework that integrates the energetic perspective of pLDDT with the topological perspective of CODE, providing a more comprehensive and reliable assessment of predicted biomolecular structures.
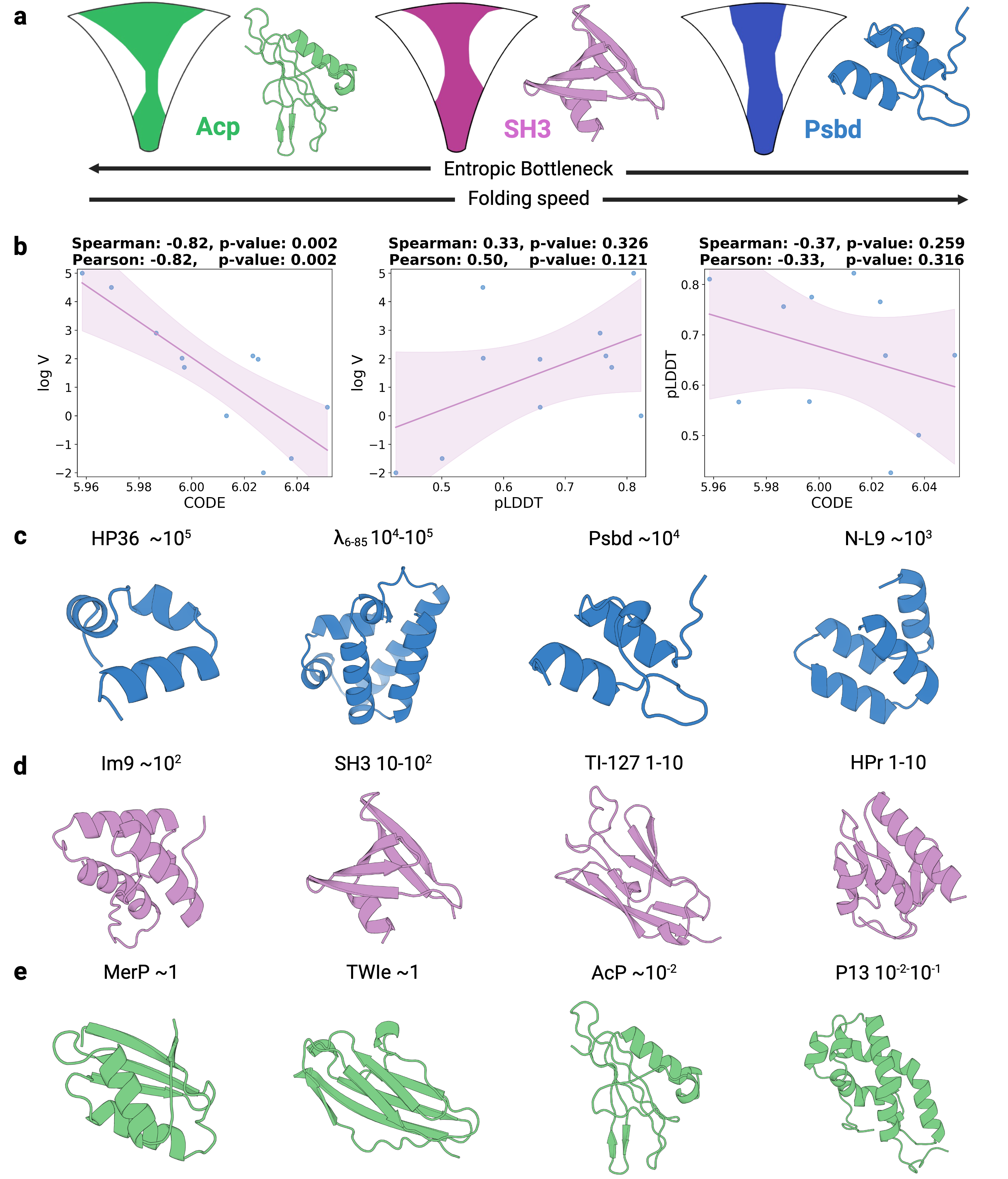
- Biology Establishes a strong empirical link between the CODE metric and protein folding rates driven by topological frustration (Spearman correlation of -0.82, p=0.002), offering a data-driven proxy for a complex biophysical phenomenon.
主要结论
- CODE demonstrates a strong, statistically significant correlation with protein folding rates mediated by topological frustration (Spearman ρ = -0.82, p=0.002), far outperforming pLDDT (ρ = 0.33, p=0.326).
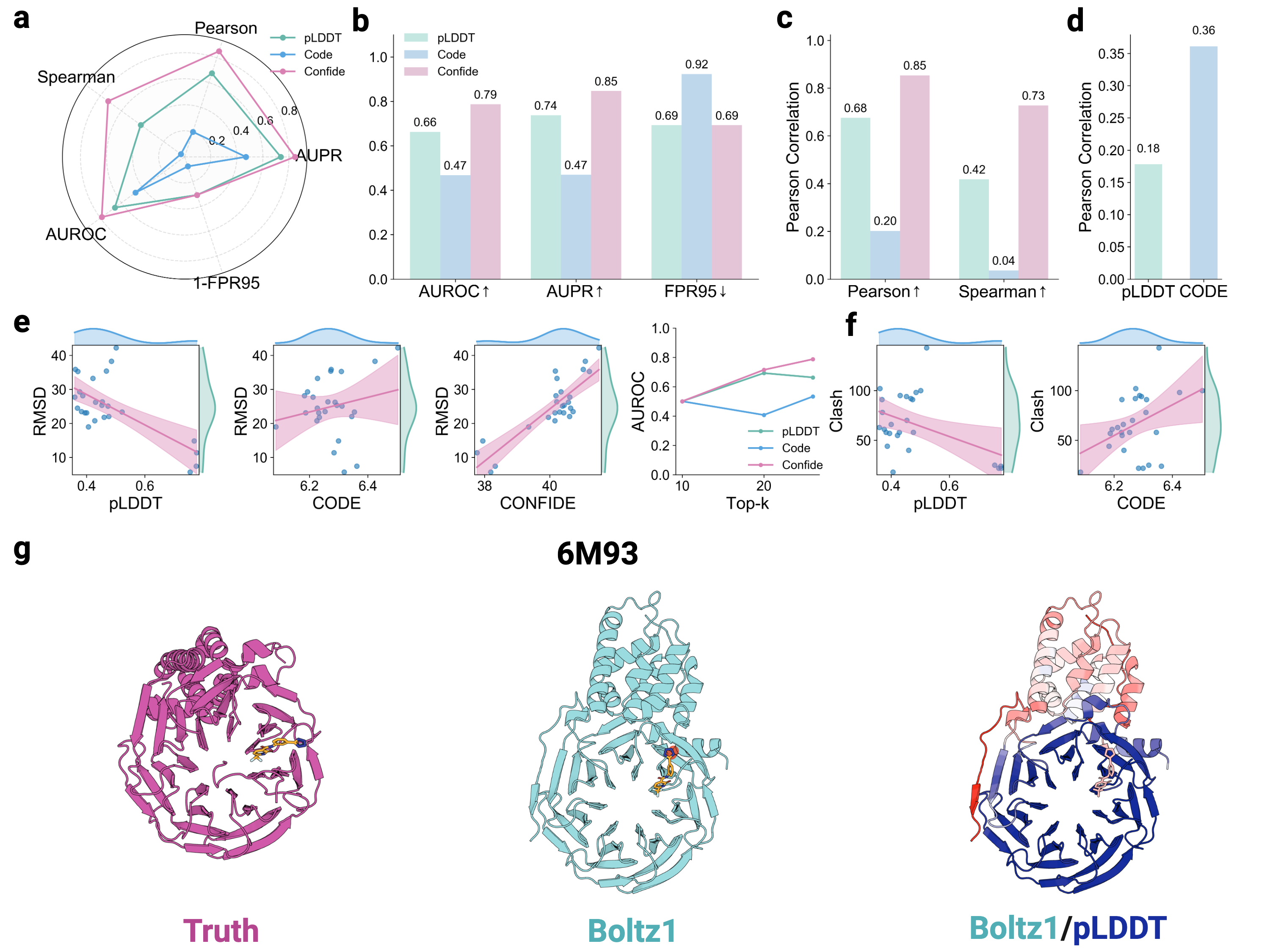
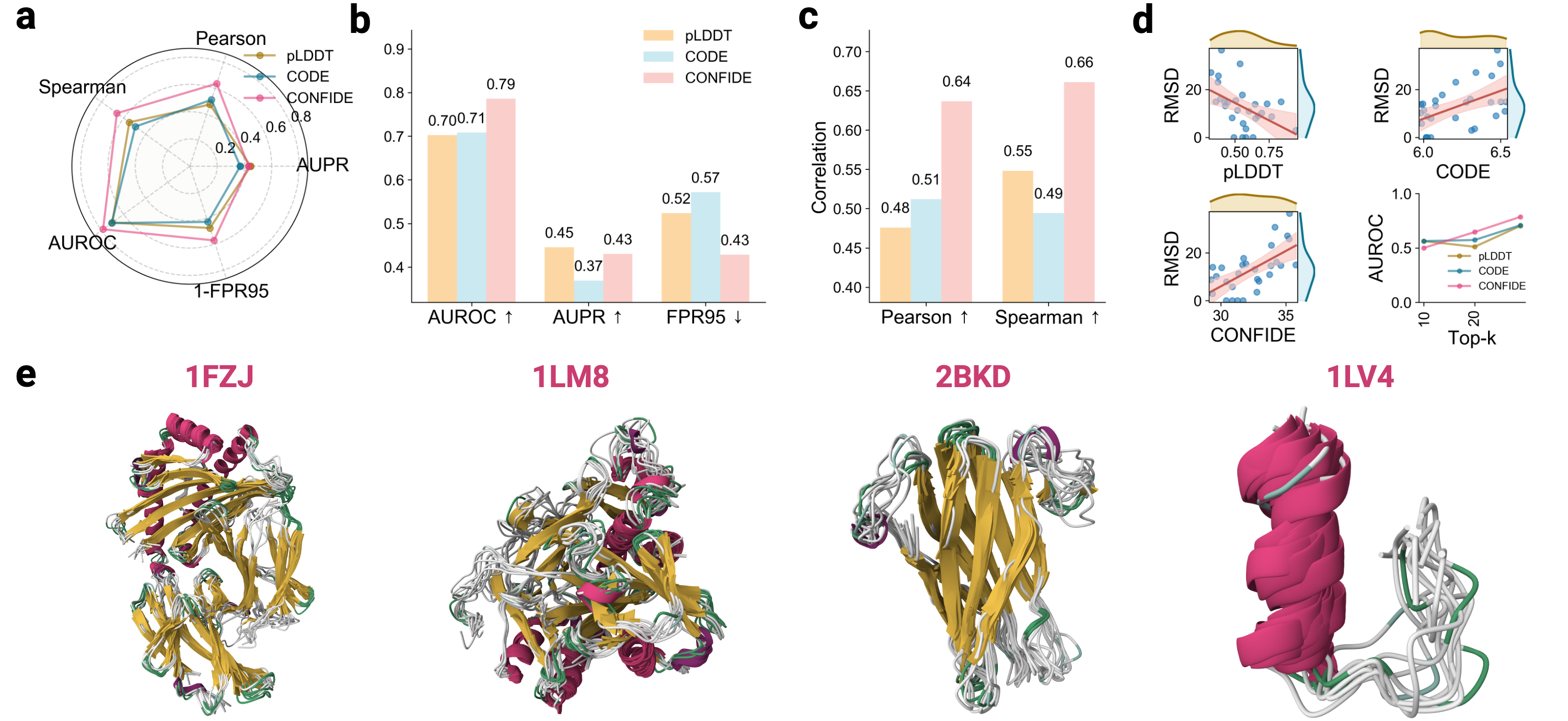
- The CONFIDE framework significantly improves hallucination detection, achieving a Spearman correlation of 0.73 with RMSD on molecular glue benchmarks, a 73.8% relative improvement over pLDDT's correlation of 0.42.
- CONFIDE enables practical downstream applications, improving binder design success rates (e.g., +13% for IAI) and accurately predicting mutation-induced binding affinity changes (Spearman ρ = 0.83 for BTK vs. Fenebrutinib, compared to pLDDT's ρ = 0.03).
摘要: Reliable evaluation of protein structure predictions remains challenging, as metrics like pLDDT capture energetic stability but often miss subtle errors such as atomic clashes or conformational traps reflecting topological frustration within the protein-folding energy landscape. We present CODE (Chain of Diffusion Embeddings), a self-evaluating metric empirically found to quantify topological frustration directly from the latent diffusion embeddings of the AlphaFold3 series of structure predictors in a fully unsupervised manner. Integrating this with pLDDT, we propose CONFIDE, a unified evaluation framework that combines energetic and topological perspectives to improve the reliability of AlphaFold3 and related models. CODE strongly correlates with protein folding rates driven by topological frustration, achieving a correlation of 0.82 compared to pLDDT’s 0.33 (a relative improvement of 148%). CONFIDE significantly enhances the reliability of quality evaluation in molecular glue structure prediction benchmarks, achieving a Spearman correlation of 0.73 with RMSD, compared to pLDDT’s correlation of 0.42, a relative improvement of 73.8%. Beyond quality assessment, our approach applies to diverse drug-design tasks, including all-atom binder design, enzymatic active-site mapping, mutation-induced binding-affinity prediction, nucleic acid aptamer screening, and flexible protein modeling. By combining data-driven embeddings with theoretical insight, CODE and CONFIDE outperform existing metrics across a wide range of biomolecular systems, offering robust and versatile tools to refine structure predictions, advance structural biology, and accelerate drug discovery.