
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
Contrastive Deep Learning for Variant Detection in Wastewater Genomic Sequencing
Georgia State University, Atlanta, Georgia, USA
30秒速读
IN SHORT: This paper addresses the core challenge of detecting viral variants in wastewater sequencing data without reference genomes or labeled annotations, overcoming issues of high noise, low coverage, and fragmented reads.
核心创新
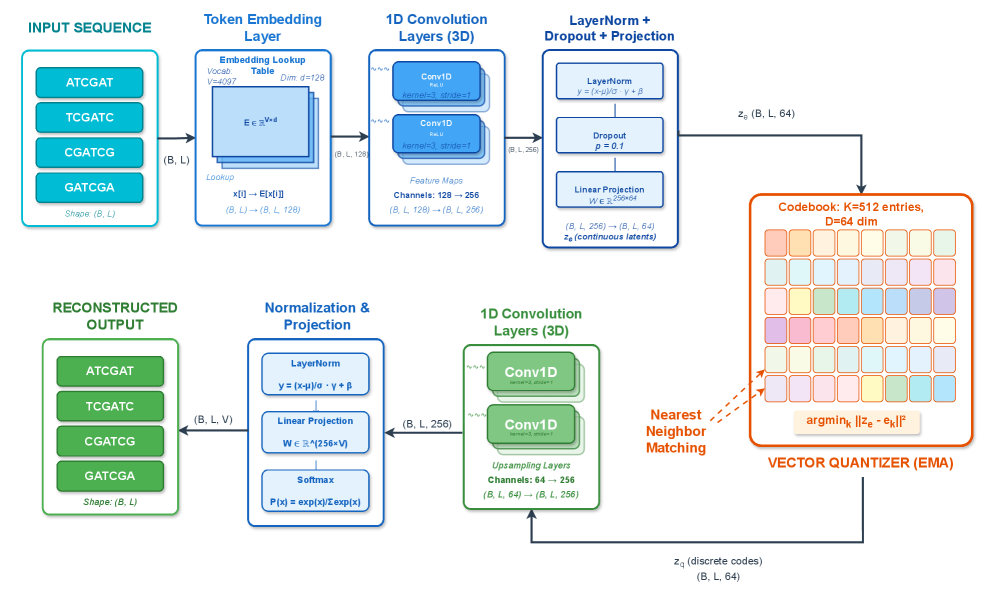
- Methodology First comprehensive application of VQ-VAE with EMA quantization to wastewater genomic surveillance, achieving 99.52% token-level reconstruction accuracy with 19.73% codebook utilization.
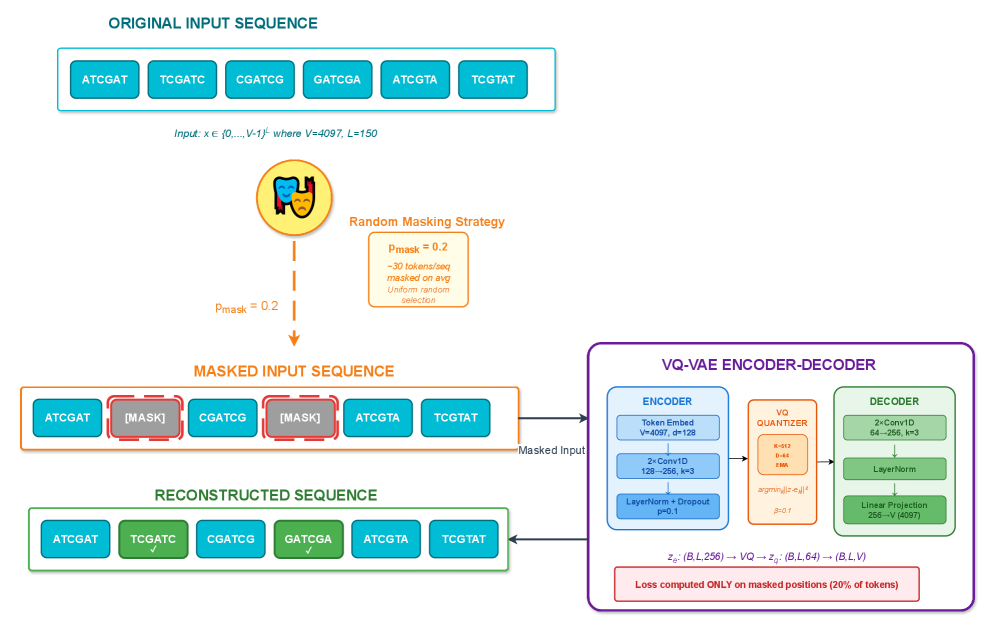
- Methodology Integration of masked reconstruction pretraining (BERT-style) maintaining ~95% accuracy under 20% token corruption, enabling robust inference with missing/low-quality data.
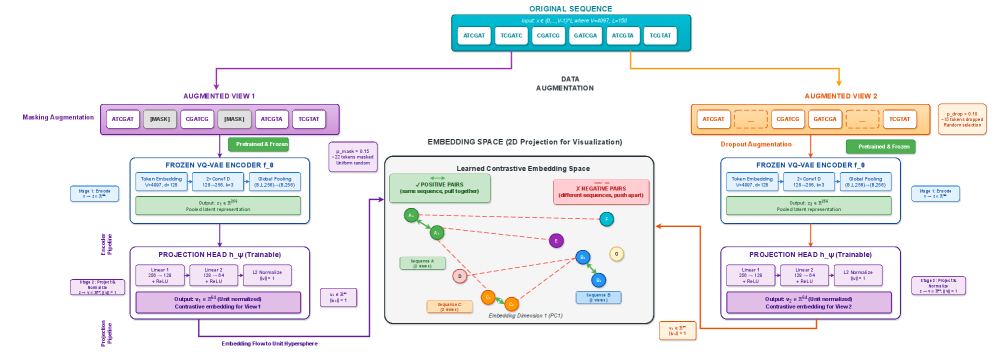
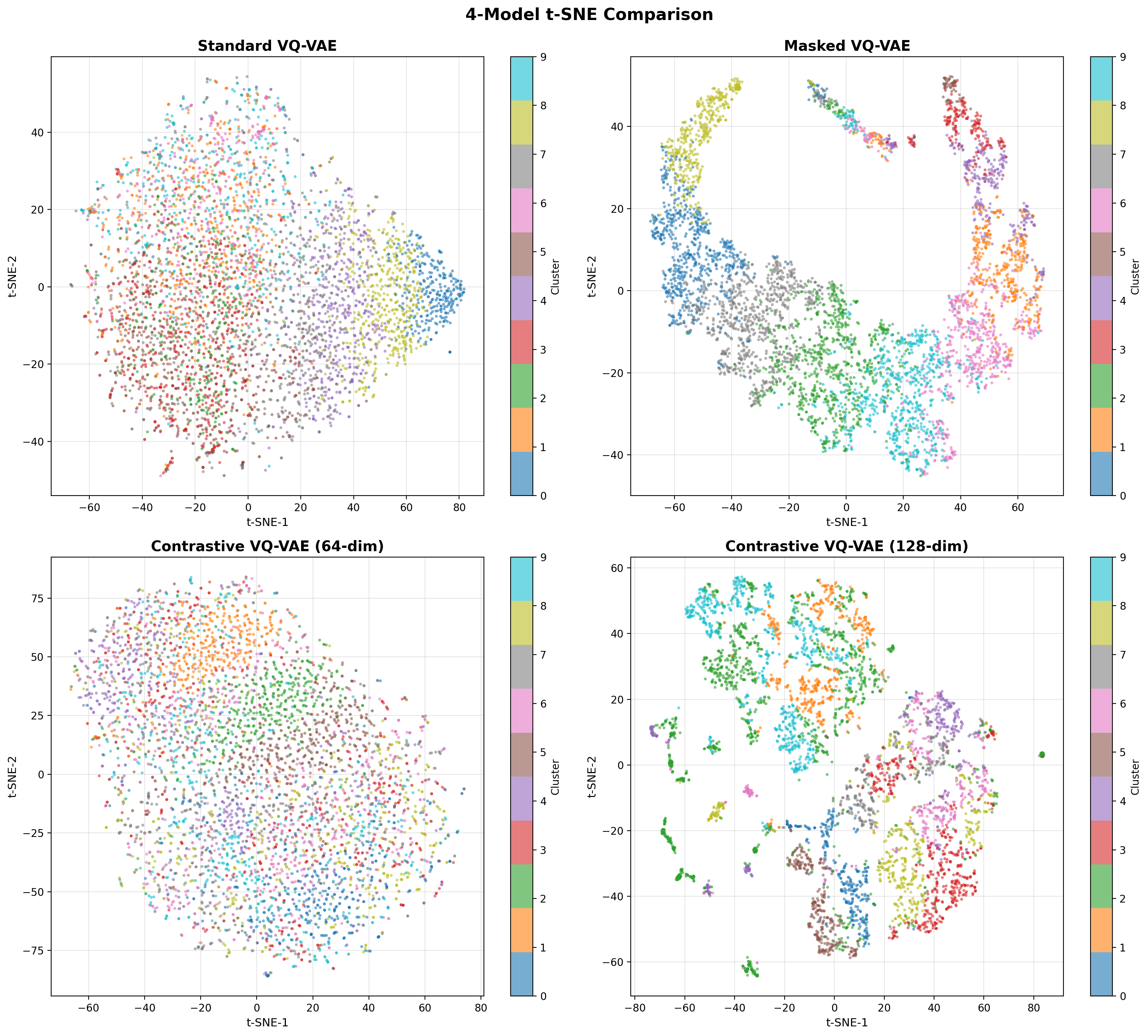
- Methodology Contrastive fine-tuning with varying embedding dimensions showing +35% (64-dim) and +42% (128-dim) Silhouette score improvements, establishing representation capacity impact on variant discrimination.
主要结论
- VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate on SARS-CoV-2 wastewater data with 100,000 reads.
- Contrastive fine-tuning improves clustering performance by +35% (0.31→0.42) with 64-dim embeddings and +42% (0.31→0.44) with 128-dim embeddings.
- The framework maintains efficient codebook utilization (19.73%, 101 of 512 codes active) while providing robust performance under data corruption.
摘要: Wastewater-based genomic surveillance has emerged as a powerful tool for population-level viral monitoring, offering comprehensive insights into circulating viral variants across entire communities. However, this approach faces significant computational challenges stemming from high sequencing noise, low viral coverage, fragmented reads, and the complete absence of labeled variant annotations. Traditional reference-based variant calling pipelines struggle with novel mutations and require extensive computational resources. We present a comprehensive framework for unsupervised viral variant detection using Vector-Quantized Variational Autoencoders (VQ-VAE) that learns discrete codebooks of genomic patterns from k-mer tokenized sequences without requiring reference genomes or variant labels. Our approach extends the base VQ-VAE architecture with masked reconstruction pretraining for robustness to missing data and contrastive learning for highly discriminative embeddings. Evaluated on SARS-CoV-2 wastewater sequencing data comprising approximately 100,000 reads, our VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate while maintaining 19.73% codebook utilization (101 of 512 codes active), demonstrating efficient discrete representation learning. Contrastive fine-tuning with different projection dimensions yields substantial clustering improvements: 64-dimensional embeddings achieve +35% Silhouette score improvement (0.31→0.42), while 128-dimensional embeddings achieve +42% improvement (0.31→0.44), clearly demonstrating the impact of embedding dimensionality on variant discrimination capability. Our reference-free framework provides a scalable, interpretable approach to genomic surveillance with direct applications to public health monitoring.