
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
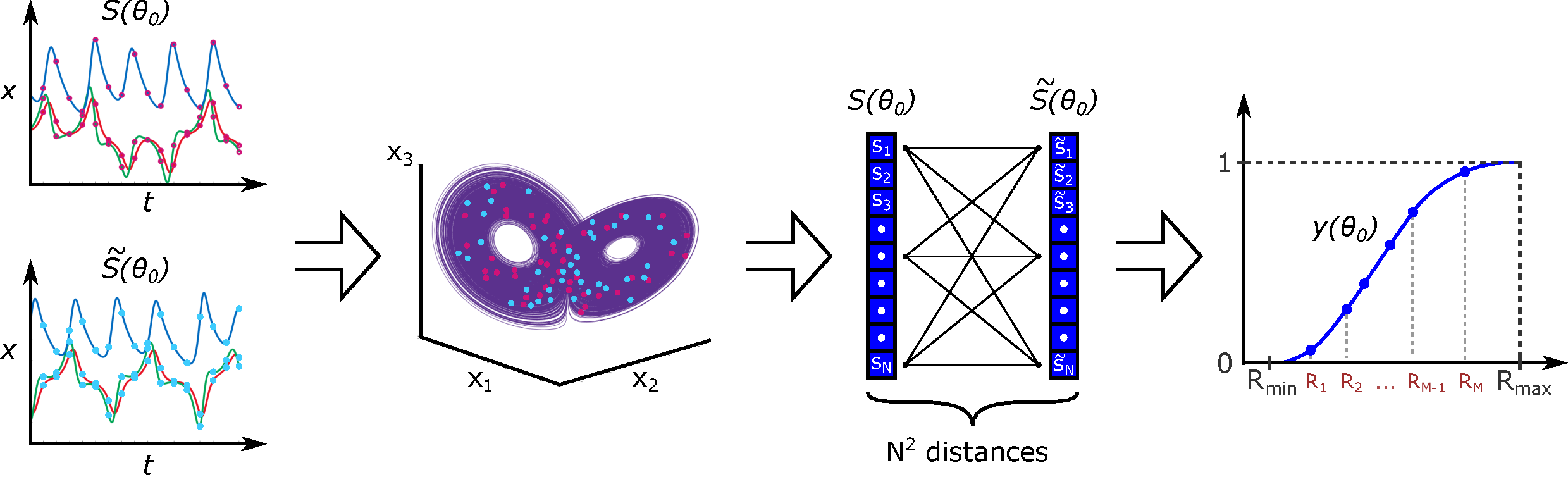
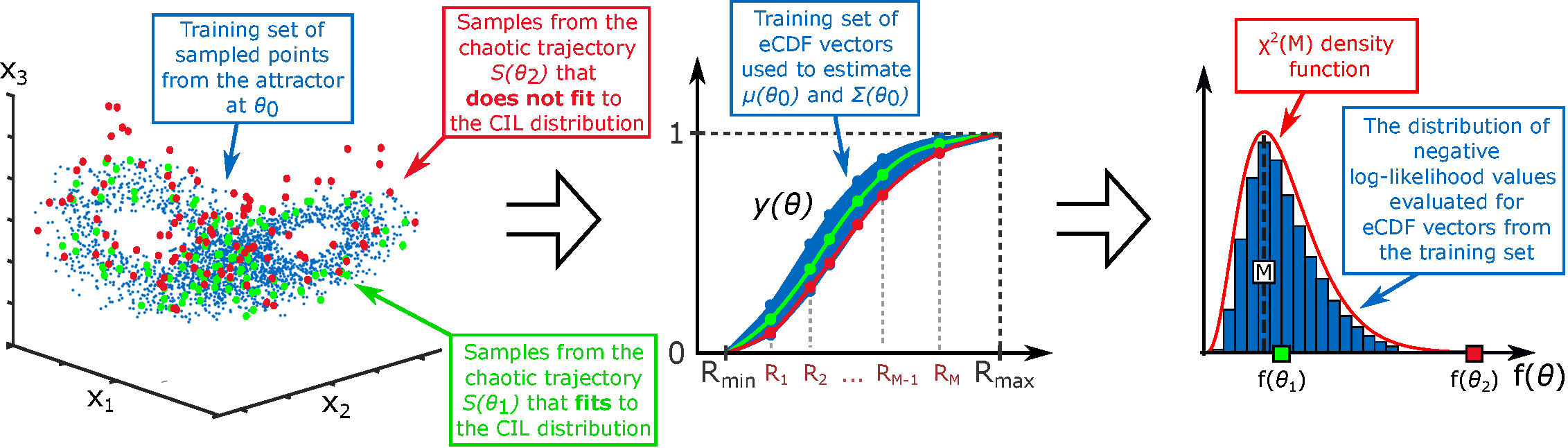
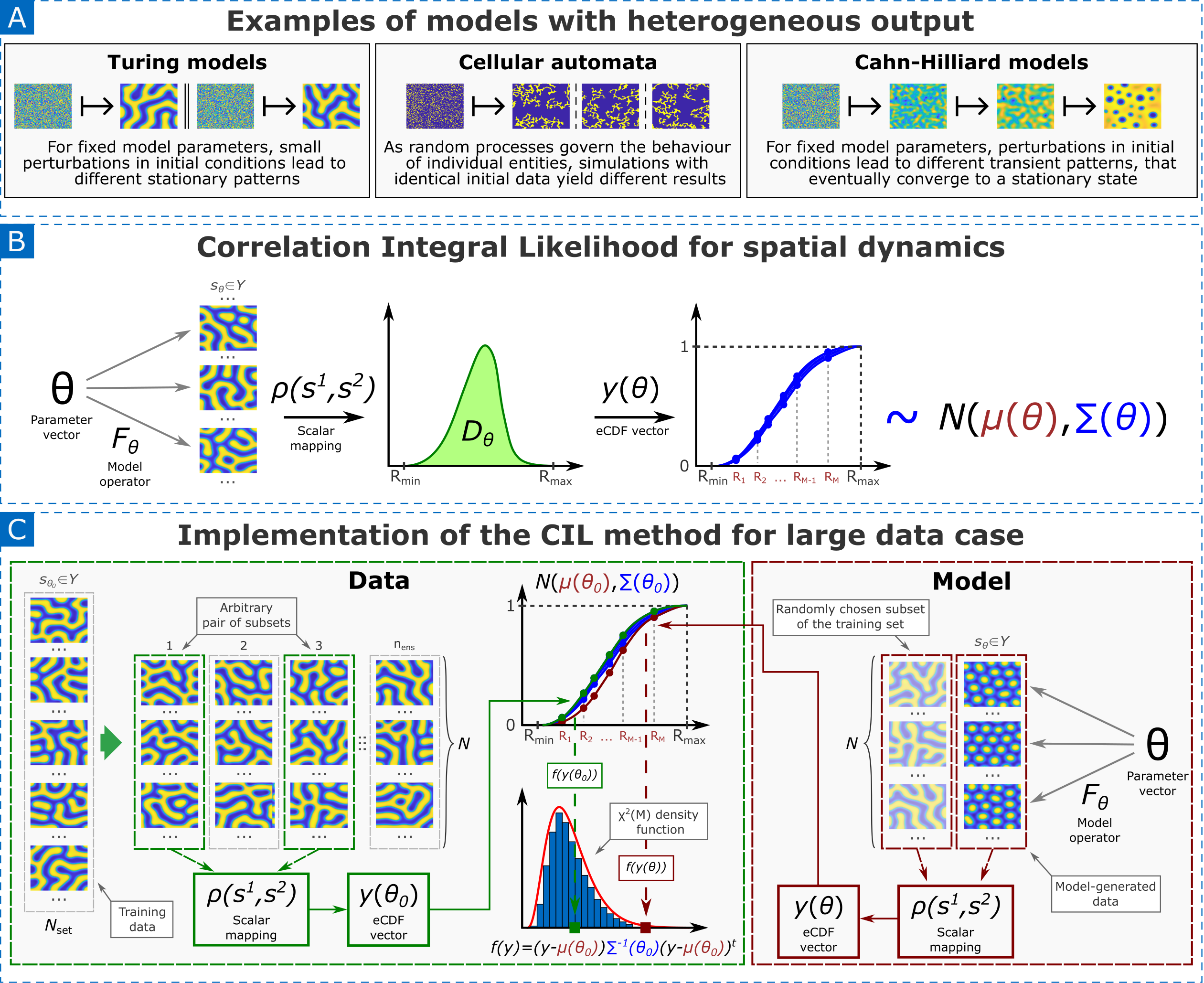
Beyond Bayesian Inference: The Correlation Integral Likelihood Framework and Gradient Flow Methods for Deterministic Sampling
Institute of Mathematics, Polish Academy of Sciences | Interdisciplinary Centre for Mathematical and Computational Modelling, University of Warsaw | Institute for Mathematics, Heidelberg University
30秒速读
IN SHORT: This paper addresses the core challenge of calibrating complex biological models (e.g., PDEs, agent-based models) with incomplete, noisy, or heterogeneous data, where traditional pointwise comparison methods fail due to system sensitivity and intrinsic variability.
核心创新
- Methodology Introduces the Correlation Integral Likelihood (CIL) framework, a unified approach for parameter estimation in systems with heterogeneous or chaotic dynamics (e.g., pattern formation, individual-based models), moving beyond classical Bayesian methods.
- Methodology Proposes integration of deterministic gradient flow methods within the CIL framework to enhance inference efficiency and accuracy, compared to traditional stochastic sampling (e.g., MCMC).
- Theory Generalizes the concept of correlation dimension from chaos theory to construct a robust metric for comparing the global geometric structure of model outputs (e.g., attractors, spatial patterns) rather than relying on unstable pointwise comparisons.
主要结论
- The CIL method provides a theoretically grounded framework for parameter estimation in systems where solution heterogeneity (e.g., in Turing patterns or chaotic attractors) makes conventional likelihoods ineffective.
- Integrating deterministic gradient flow sampling with the CIL framework can potentially enhance computational efficiency and inference accuracy compared to purely stochastic methods like MCMC, especially for high-dimensional parameter spaces.
- The approach enables reliable model calibration and validation even with incomplete, noisy, or single-snapshot data, advancing the predictive capability and mechanistic understanding of complex biological systems.
摘要: Calibrating mathematical models of biological processes is essential for achieving predictive accuracy and gaining mechanistic insight. However, this task remains challenging due to limited and noisy data, significant biological variability, and the computational complexity of the models themselves. In this method's article, we explore a range of approaches for parameter inference in partial differential equation (PDE) models of biological systems. We introduce a unified mathematical framework, the Correlation Integral Likelihood (CIL) method, for parameter estimation in systems exhibiting heterogeneous or chaotic dynamics, encompassing both pattern formation models and individual-based models. Departing from classical Bayesian inverse problem methodologies, we motivate the development of the CIL method, demonstrate its versatility, and highlight illustrative applications within mathematical biology. Furthermore, we compare stochastic sampling strategies, such as Markov Chain Monte Carlo (MCMC), with deterministic gradient flow approaches, highlighting how these methods can be integrated within the proposed framework to enhance inference performance. Our work provides a practical and theoretically grounded toolbox for researchers seeking to calibrate complex biological models using incomplete, noisy, or heterogeneous data, thereby advancing both the predictive capability and mechanistic understanding of such systems.