
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
Simulation and inference methods for non-Markovian stochastic biochemical reaction networks
School of Mathematical Sciences, Queensland University of Technology | Centre for Data Science, Queensland University of Technology | ARC Centre of Excellence for Mathematical Analysis of Cellular Systems (MACSYS), Queensland University of Technology
30秒速读
IN SHORT: This paper addresses the computational bottleneck of simulating and performing Bayesian inference for non-Markovian biochemical systems with history-dependent delays, which are crucial for modeling processes like gene transcription but are prohibitively expensive with existing methods.
核心创新
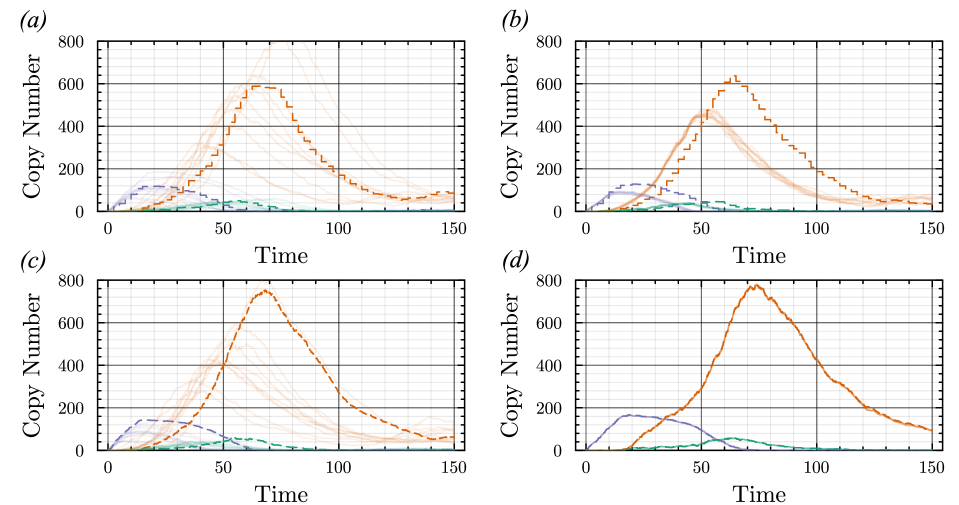
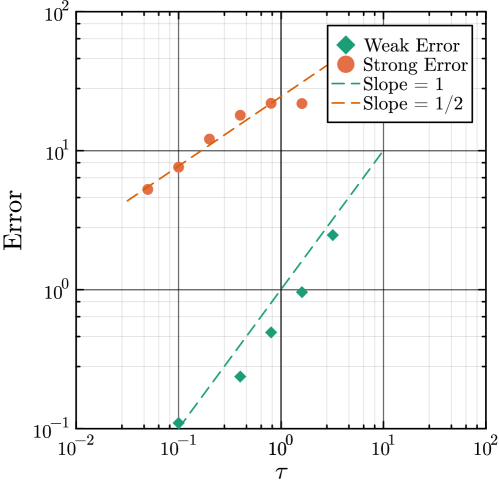
- Methodology Generalizes the next reaction method and τ-leaping to support arbitrary inter-event time distributions for non-Markovian systems, maintaining computational scalability.
- Methodology Introduces a novel coupling scheme to generate positively correlated exact and approximate non-Markovian sample paths, a prerequisite for variance reduction techniques.
- Methodology Enables the application of multifidelity and multilevel Monte Carlo (MLMC) methods to non-Markovian systems for the first time, bridging a significant methodological gap.
主要结论
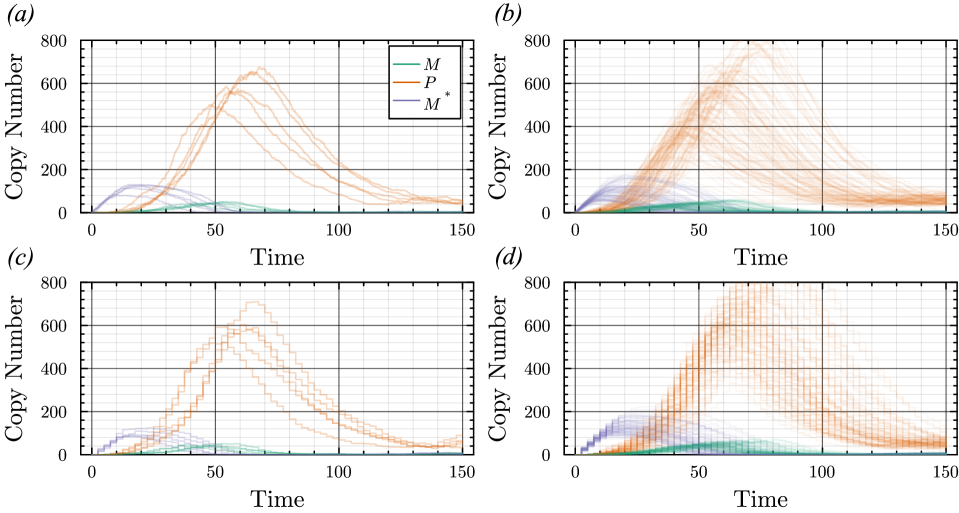
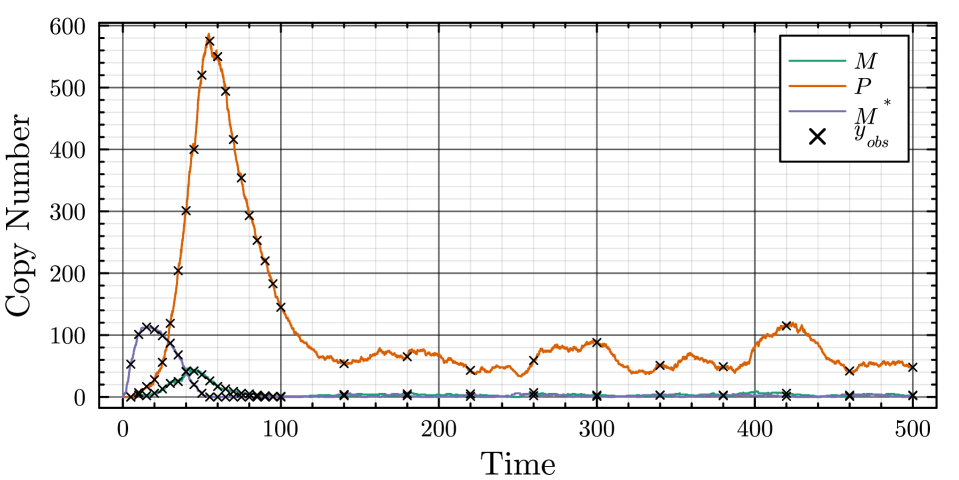
- The proposed non-Markovian simulation algorithms and coupling scheme successfully enable multifidelity inference, demonstrated on a gene regulation model with delayed auto-inhibition.
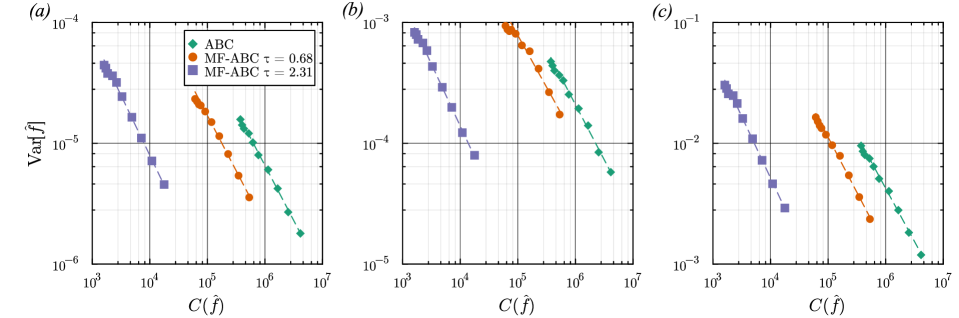
- The method achieves a computational speedup of two orders of magnitude (100x) in inference efficiency compared to standard approaches for the non-Markovian case study.
- The framework supports arbitrary delay distributions (state- and time-dependent), significantly extending the practical modeling scope beyond previous methods limited to simpler, time-only delays.
摘要: Stochastic models of biochemical reaction networks are widely used to capture intrinsic noise in cellular systems. The typical formulation of these models are based on Markov processes for which there is extensive research on efficient simulation and inference. However, there are biological processes, such as gene transcription and translation, that introduce history dependent dynamics requiring non-Markovian processes to accurately capture the stochastic dynamics of the system. This greater realism comes with additional computational challenges for simulation and parameter inference. We develop efficient stochastic simulation algorithms for well-mixed non-Markovian stochastic biochemical reaction networks with delays that depend on system state and time. Our methods generalize the next reaction method and τ-leaping method to support arbitrary inter-event time distributions while preserving computational scalability. We also introduce a coupling scheme to generate exact non-Markovian sample paths that are positively correlated to an approximate non-Markovian τ-leaping sample path. This enables substantial computational gains for Bayesian inference of model parameters though multifidelity simulation-based inference schemes. We demonstrate the effectiveness of our approach on a gene regulation model with delayed auto-inhibition, showing substantial gains in both simulation accuracy and inference efficiency of two orders of magnitude. These results extend the practical applicability of non-Markovian models in systems biology and beyond.