
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
Department of Computer Science and Genome Center, University of California, Davis | Architecture et Dynamique des Macromolécules Biologiques, UMR 3528 du CNRS, Institut Pasteur | Department of Physics, School of Sciences, Great Bay University | Université Paris-Saclay, CNRS, CEA, Institut de Physique Théorique
30秒速读
IN SHORT: This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein conformations, a problem where existing methods often produce non-physical trajectories due to oversimplified energy surfaces and steric clashes.
核心创新
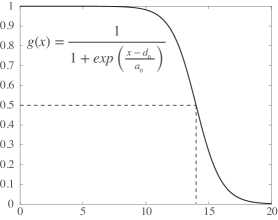
- Methodology Introduces SIDE (Stochastic Integro-Differential Equation), a novel Langevin bridge-based framework that efficiently approximates exact bridge equations at low temperatures to generate constrained transition trajectories.
- Methodology Develops a new coarse-grained potential that combines a Gō-like term (to preserve native backbone geometry) with a Rouse-type elastic energy term (from polymer physics), avoiding the problematic mixing of start/target conformation information used in prior methods like MinActionPath.
- Theory Provides a rigorous stochastic integro-differential formulation derived from the Langevin bridge formalism, which explicitly constrains trajectories to reach a target state within finite time, moving beyond Minimum Action Path (MAP) principles.
主要结论
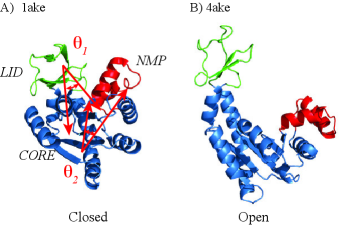
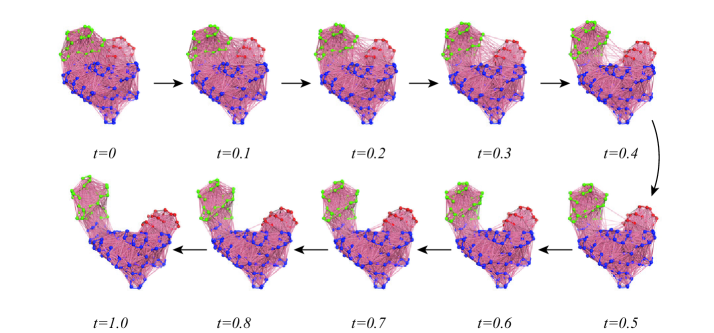
- The SIDE framework generates smooth, low-energy transition trajectories that maintain realistic molecular geometry, as demonstrated on several proteins undergoing large-scale conformational changes.
- SIDE frequently recovers experimentally supported intermediate states along transition paths, suggesting its paths have biological relevance beyond mere endpoint interpolation.
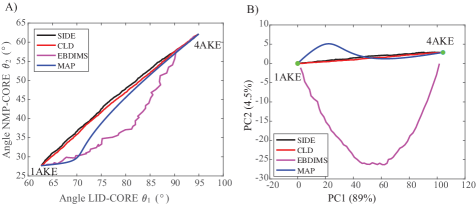
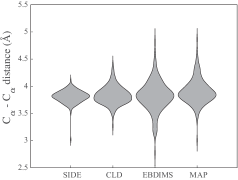
- Compared to established methods like MinActionPath and EBDIMS, SIDE offers improved physical realism and computational efficiency for modeling biomolecular conformational transitions, though challenges remain for highly complex motions.
摘要: We introduce a computational framework for generating realistic transition paths between distinct conformations of large biomolecular systems. The method is built on a stochastic integro-differential formulation derived from the Langevin bridge formalism, which constrains molecular trajectories to reach a prescribed final state within a finite time and yields an efficient low-temperature approximation of the exact bridge equation. To obtain physically meaningful protein transitions, we couple this formulation to a new coarse-grained potential combining a Gō-like term that preserves native backbone geometry with a Rouse-type elastic energy term from polymer physics; we refer to the resulting approach as SIDE. We evaluate SIDE on several proteins undergoing large-scale conformational changes and compare its performance with established methods such as MinActionPath and EBDIMS. SIDE generates smooth, low-energy trajectories that maintain molecular geometry and frequently recover experimentally supported intermediate states. Although challenges remain for highly complex motions—largely due to the simplified coarse-grained potential—our results demonstrate that SIDE offers a powerful and computationally efficient strategy for modeling biomolecular conformational transitions.