
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
Incorporating indel channels into average-case analysis of seed-chain-extend
Carnegie Mellon University, Pittsburgh, PA, USA
30秒速读
IN SHORT: This paper addresses the core pain point of bridging the theoretical gap for the widely used seed-chain-extend heuristic by providing the first rigorous average-case analysis that accounts for insertions and deletions (indels), not just substitutions.
核心创新
- Methodology Introduces a generalized definition of 'recoverability' and a 'homologous path' to mathematically model the correct alignment under indel mutation channels, moving beyond the simpler 'homologous diagonal' used for substitutions only.
- Theory Develops new mathematical machinery to handle the dependence structure of neighboring anchors and the existence of 'clipping anchors' (partially correct anchors), which are unique challenges introduced by indels.
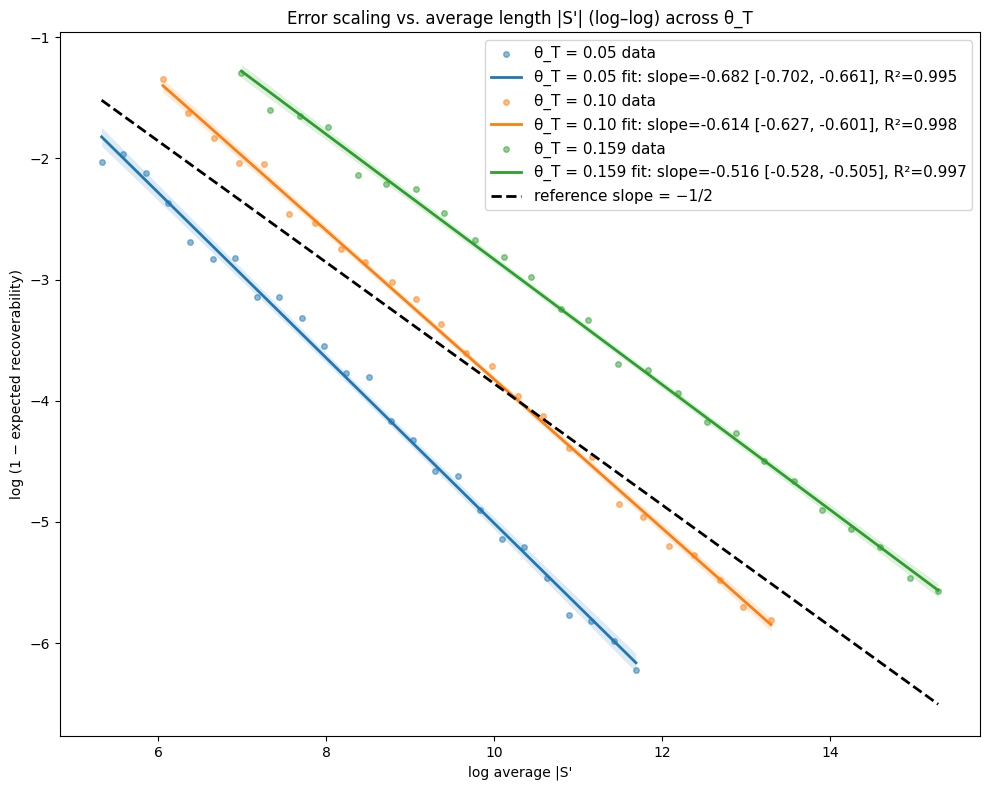
- Theory Proves that under a total mutation rate θ_T < 0.159, optimal linear-gap cost chaining achieves an expected recoverability of ≥ 1 - O(1/√m), generalizing the prior substitution-only result to a biologically realistic model.
主要结论
- The expected recoverability of an optimal chain under linear-gap cost chaining is ≥ 1 - O(1/√m) when the total mutation rate θ_T (sum of substitution, insertion, deletion rates) is less than 0.159.
- The expected runtime of the algorithm is O(m n^(3.15·θ_T) log n). For example, at a θ_T of 0.05 (similar to human-chimp divergence), the exponent is ~1.12, leading to near-linear scaling.
- The analysis successfully bridges theory and practice by extending the proof framework to handle indels, justifying the heuristic's empirical effectiveness on real genomic data which contains indels.
摘要: Given a sequence s1 of n letters drawn i.i.d. from an alphabet of size σ and a mutated substring s2 of length m<n, we often want to recover the mutation history that generated s2 from s1. Modern sequence aligners are widely used for this task, and many employ the seed-chain-extend heuristic with k-mer seeds. Previously, Shaw and Yu showed that optimal linear-gap cost chaining can produce a chain with 1−O(1/m) recoverability, the proportion of the mutation history that is recovered, in O(mn^(2.43θ) log n) expected time, where θ<0.206 is the mutation rate under a substitution-only channel and s1 is assumed to be uniformly random. However, a gap remains between theory and practice, since real genomic data includes insertions and deletions (indels), and yet seed-chain-extend remains effective. In this paper, we generalize those prior results by introducing mathematical machinery to deal with the two new obstacles introduced by indel channels: the dependence of neighboring anchors and the presence of anchors that are only partially correct. We are thus able to prove that the expected recoverability of an optimal chain is ≥1−O(1/√m) and the expected runtime is O(mn^(3.15·θ_T) log n), when the total mutation rate given by the sum of the substitution, insertion, and deletion mutation rates (θ_T = θ_i + θ_d + θ_s) is less than 0.159.