
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
scCluBench: Comprehensive Benchmarking of Clustering Algorithms for Single-Cell RNA Sequencing
Not specified in provided content
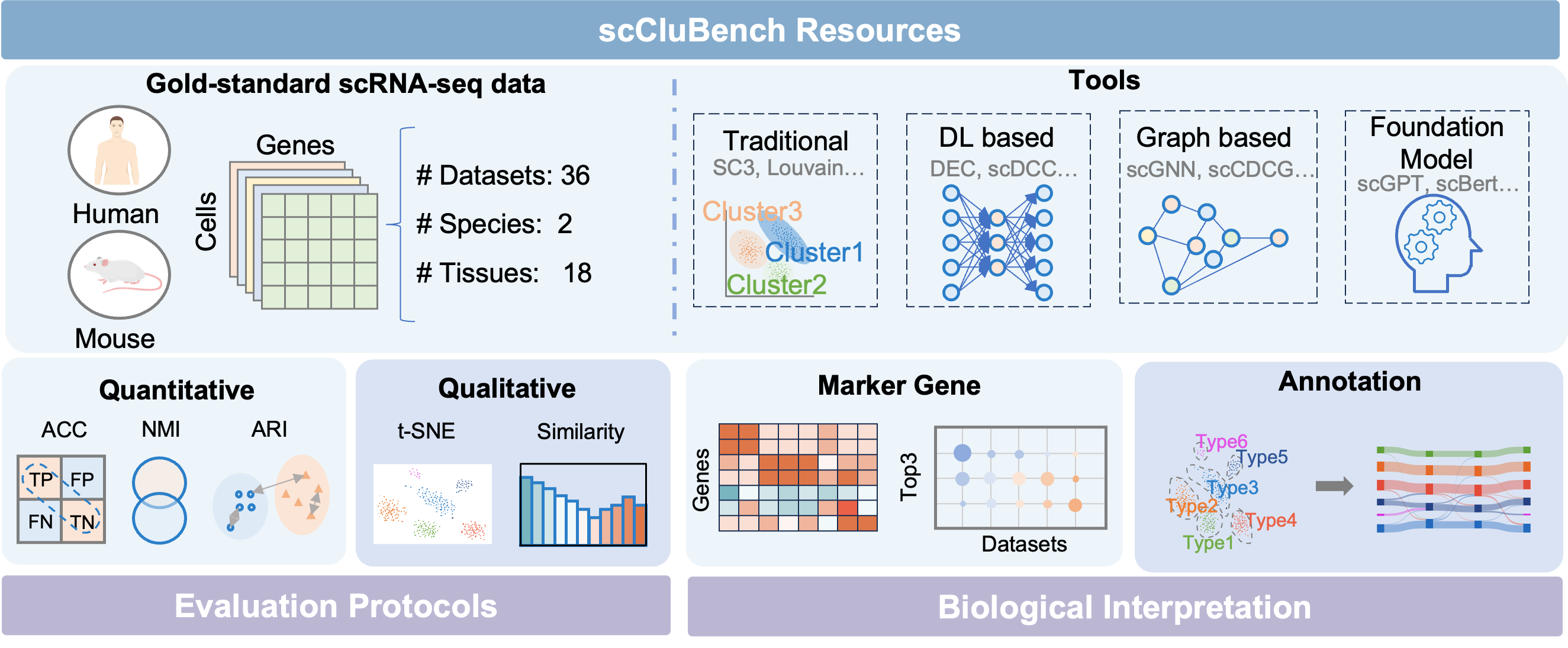
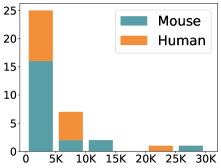
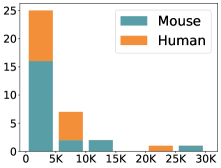
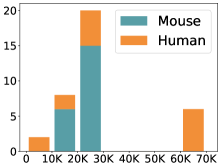
30秒速读
IN SHORT: This paper addresses the critical gap of fragmented and non-standardized benchmarking in single-cell RNA-seq clustering, which hinders objective comparison and selection of appropriate methods for specific biological contexts.
核心创新
- Methodology Introduces scCluBench, the first comprehensive benchmarking framework that systematically evaluates 16 clustering methods across four categories (traditional, deep learning-based, graph-based, and foundation models) on 36 standardized datasets.
- Methodology Establishes standardized protocols for biological interpretation, including reproducible pipelines for marker gene identification and two distinct cell type annotation approaches (best-mapping and marker-overlap), validated with gold-standard references.
- Methodology Provides a unified and modular benchmarking workflow covering data preprocessing, clustering, and annotation with standardized input-output formats, ensuring reproducibility and fair comparison.
主要结论
- scCDCG (a cut-informed graph embedding model) achieved the highest average clustering accuracy (81.29 ± 1.45) across 36 datasets, outperforming other graph-based, deep learning, and traditional methods.
- Biological foundation models (scGPT, GeneFormer, GeneCompass) showed strong performance in classification tasks (e.g., scGPT achieved 98.14% ACC on Sapiens Ear Crista Ampullaris) but underperformed in direct clustering, highlighting a trade-off between general representation and task-specific optimization.
- The benchmark reveals method-specific limitations: traditional methods struggle with sparse data, deep learning models may fail to capture cell relationships, and graph-based models can suffer from over-smoothing, while most methods decouple embedding learning from clustering optimization.
摘要: Cell clustering is crucial for uncovering cellular heterogeneity in single-cell RNA sequencing (scRNA-seq) data by identifying cell types and marker genes. Despite its importance, benchmarks for scRNA-seq clustering methods remain fragmented, often lacking standardized protocols and failing to incorporate recent advances in artificial intelligence. To fill these gaps, we present scCluBench, a comprehensive benchmark of clustering algorithms for scRNA-seq data. First, scCluBench provides 36 scRNA-seq datasets collected from diverse public sources, covering multiple tissues, which are uniformly processed and standardized to ensure consistency for systematic evaluation and downstream analyses. To evaluate performance, we collect and reproduce a range of scRNA-seq clustering methods, including traditional, deep learning-based, graph-based, and biological foundation models. We comprehensively evaluate each method both quantitatively and qualitatively, using core performance metrics as well as visualization analyses. Furthermore, we construct representative downstream biological tasks, such as marker gene identification and cell type annotation, to further assess the practical utility. scCluBench then investigates the performance differences and applicability boundaries of various clustering models across diverse analytical tasks, systematically assessing their robustness and scalability in real-world scenarios. Overall, scCluBench offers a standardized and user-friendly benchmark for scRNA-seq clustering, with curated datasets, unified evaluation protocols, and transparent analyses, facilitating informed method selection and providing valuable insights into model generalizability and application scope.222All datasets, code, and the Extended version for scCluBench are available at the link: https://github.com/XPgogogo/scCluBench. More details for each stage are provided in the extended version.