
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
On the Approximation of Phylogenetic Distance Functions by Artificial Neural Networks
Indiana University, Bloomington, IN 47405, USA
30秒速读
IN SHORT: This paper addresses the core challenge of developing computationally efficient and scalable neural network architectures that can learn accurate phylogenetic distance functions from simulated data, bridging the gap between simple distance methods and complex model-based inference.
核心创新
- Methodology Introduces minimal, permutation-invariant neural architectures (Sequence networks S and Pair networks P) specifically designed to approximate phylogenetic distance functions, ensuring invariance to taxa ordering without costly data augmentation.
- Methodology Leverages theoretical results from metric embedding (Bourgain's theorem, Johnson-Lindenstrauss Lemma) to inform network design, explicitly linking embedding dimension to the number of taxa for efficient representation.
- Methodology Demonstrates how equivariant layers and attention mechanisms can be structured to handle both i.i.d. and spatially correlated sequence data (e.g., models with indels or rate variation), adapting to the complexity of the generative evolutionary model.
主要结论
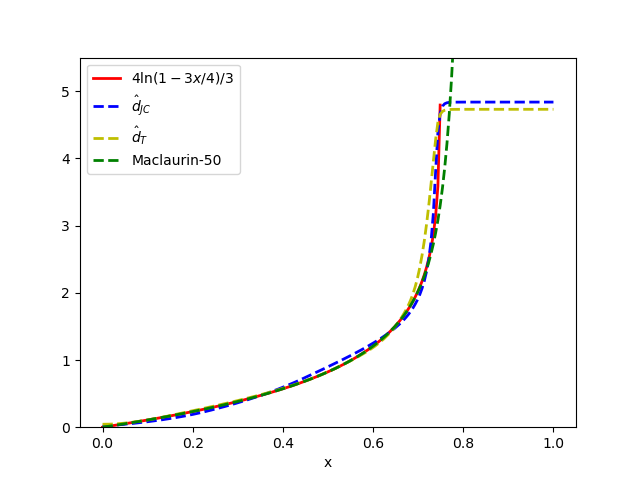
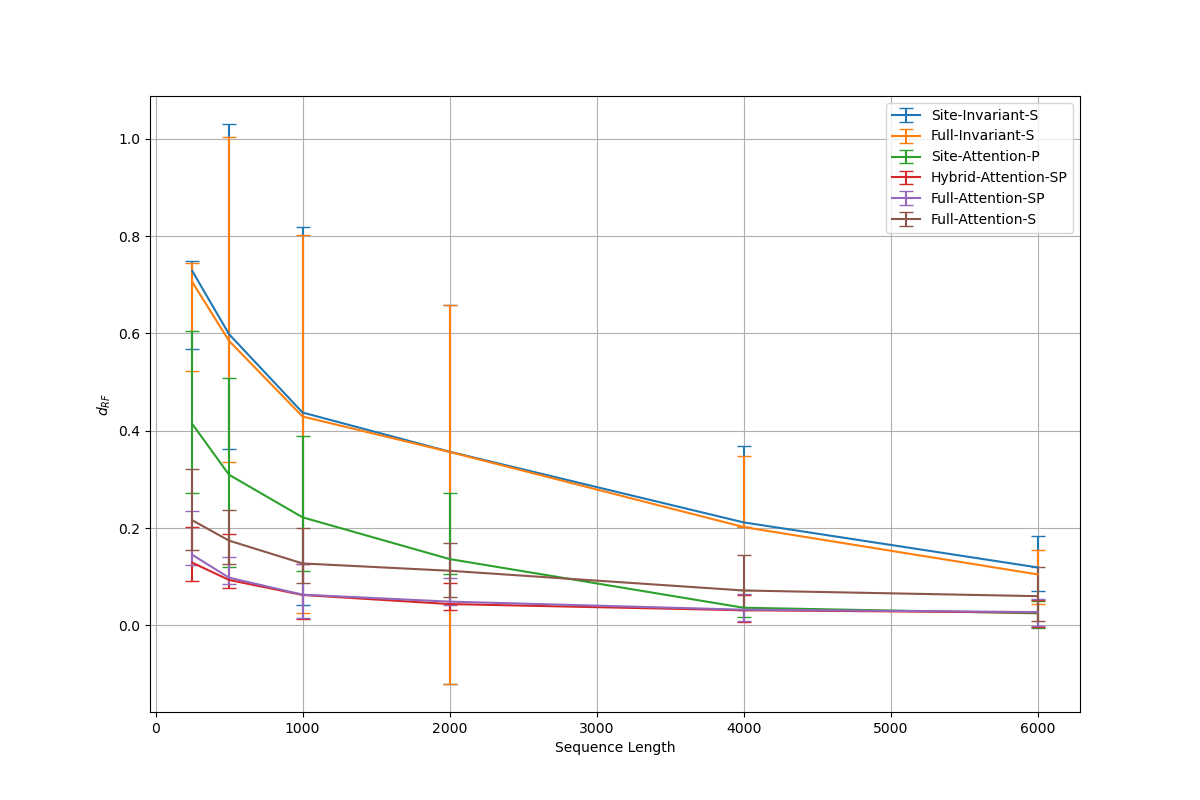
- The proposed minimal architectures (e.g., Sites-Invariant-S with ~7.6K parameters) achieve results comparable to state-of-the-art inference methods like IQ-TREE on simulated data under various models (JC, K2P, HKY, LG+indels), outperforming classic pairwise distance methods (d_H, d_JC, d_K2P) in most conditions.
- Architectures incorporating taxa-wise attention, while more memory-intensive, are necessary for complex evolutionary models with spatial dependencies; however, simpler networks suffice for simpler i.i.d. models, indicating an architecture-evolutionary model correspondence.
- Performance is highly sensitive to hyperparameters: validation error increases sharply with fewer than 4 attention heads or with hidden channel counts outside an optimal range (e.g., 32-128), aligning with theoretical requirements for learning graph-structured data.
摘要: Inferring the phylogenetic relationships among a sample of organisms is a fundamental problem in modern biology. While distance-based hierarchical clustering algorithms achieved early success on this task, these have been supplanted by Bayesian and maximum likelihood search procedures based on complex models of molecular evolution. In this work we describe minimal neural network architectures that can approximate classic phylogenetic distance functions and the properties required to learn distances under a variety of molecular evolutionary models. In contrast to model-based inference (and recently proposed model-free convolutional and transformer networks), these architectures have a small computational footprint and are scalable to large numbers of taxa and molecular characters. The learned distance functions generalize well and, given an appropriate training dataset, achieve results comparable to state-of-the art inference methods.