
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
GOPHER: Optimization-based Phenotype Randomization for Genome-Wide Association Studies with Differential Privacy
Department of Biomedical Informatics & Data Science, Yale School of Medicine | Department of Technology and Operations Management, Harvard Business School | Department of Computer Science, Yale University
30秒速读
IN SHORT: This paper addresses the core challenge of balancing rigorous privacy protection with data utility when releasing full GWAS summary statistics, overcoming the limitations of prior methods that either add excessive noise or restrict output to a small subset of results.
核心创新
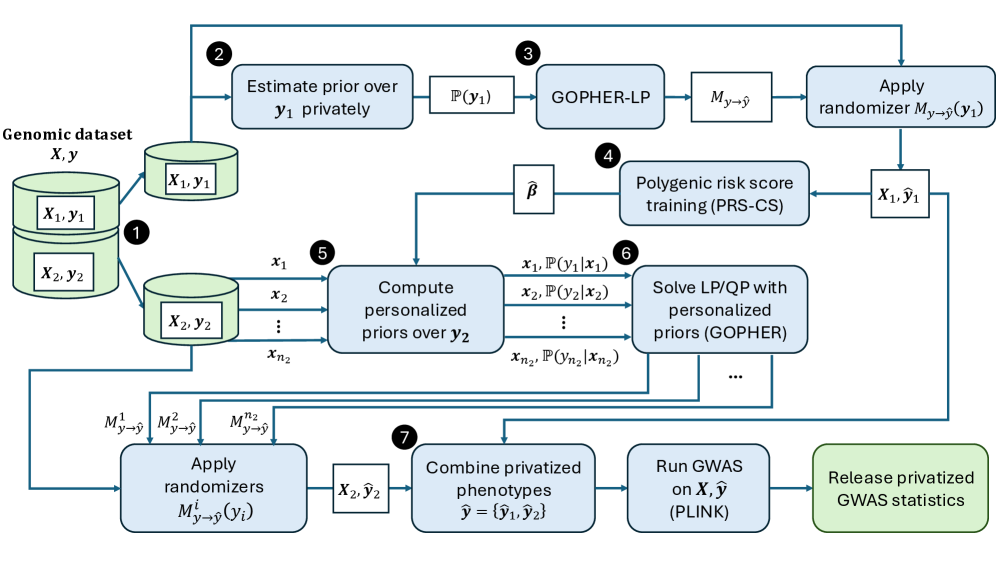
- Methodology Introduces an optimization-based phenotype randomization mechanism (GOPHER-LP) that directly minimizes expected error in GWAS statistics, formulated as a linear programming problem to enhance utility beyond baseline methods like randomized response.
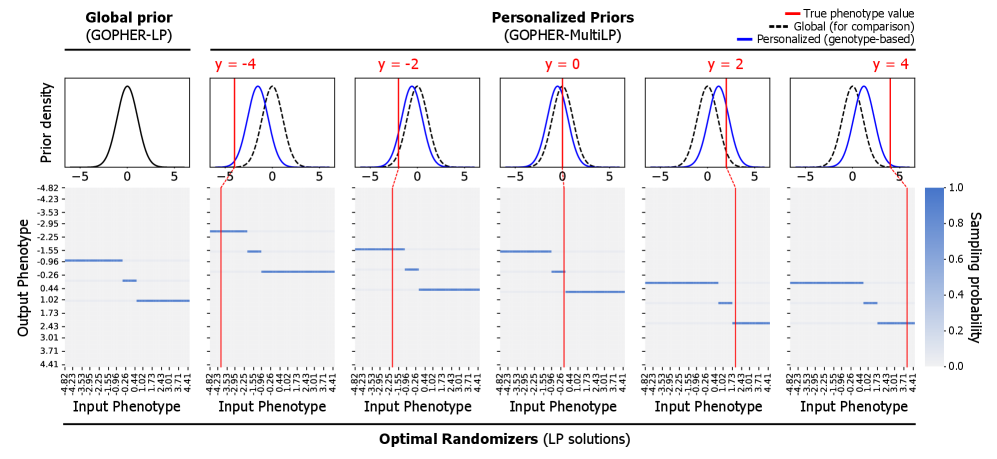
- Methodology Proposes GOPHER-MultiLP, which incorporates personalized priors derived from predictive models (e.g., polygenic risk scores) trained on a held-out subset, enabling sample-specific optimization that leverages genotype information to further reduce noise.
- Theory Adopts and extends the concept of phenotypic differential privacy (analogous to label DP), focusing protection on sensitive phenotypes while treating genotypes as public, providing a practical middle ground between full DP and unrestricted release.
主要结论
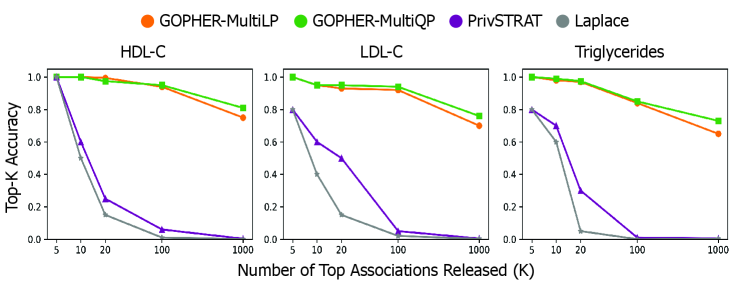
- The GOPHER framework enables the release of complete GWAS statistics (e.g., over 500,000 variants) with provable privacy guarantees, a significant scalability advance over prior methods limited to releasing only 3-5 top associations.
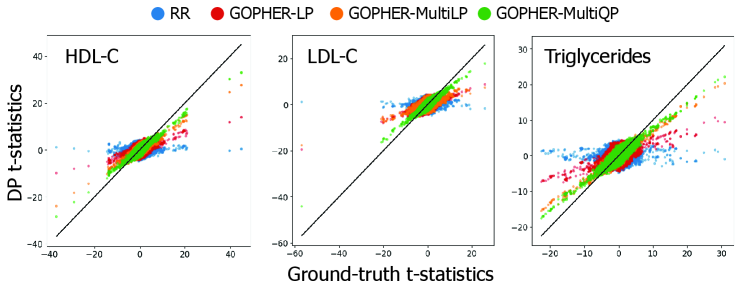
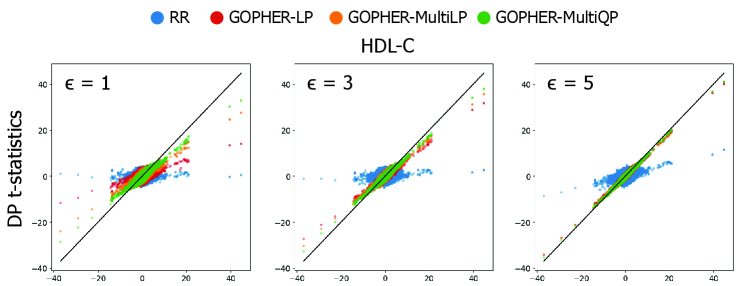
- Experiments on UK Biobank data (n=100,000) demonstrate that the mechanisms yield association statistics that accurately match non-private GWAS results while maintaining rigorous (ε, δ)-DP guarantees.
- The phenotype-randomization approach decouples the added noise from the number of genetic variants analyzed, addressing a fundamental scalability challenge not previously solved in the DP-GWAS literature.
摘要: Genome-wide association studies (GWAS) are an essential tool in biomedical research for identifying genetic factors linked to health and disease. However, publicly releasing GWAS summary statistics poses well-recognized privacy risks, including the potential to infer an individual’s participation in the study or to reveal sensitive phenotypic information (e.g., disease status). While differential privacy (DP) offers a rigorous mathematical framework for mitigating these risks, existing DP techniques for GWAS either introduce excessive noise or restrict the release to a limited set of results. In this work, we present practical DP mechanisms for releasing the complete set of genome-wide association statistics with privacy guarantees. We demonstrate the accuracy of the privacy-preserving statistics released by our mechanisms on a range of GWAS datasets from the UK Biobank, utilizing both real and simulated phenotypes. We introduce two key techniques to overcome the limitations of prior approaches: (1) an optimization-based randomization mechanism that directly minimizes the expected error in GWAS results to enhance utility, and (2) the use of personalized priors, derived from predictive models privately trained on a subset of the dataset, to enable sample-specific optimization which further reduces the amount of noise introduced by DP. Overall, our work provides practical tools for accurately releasing comprehensive GWAS results with provable protection of study participants.