
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
Hierarchical Molecular Language Models (HMLMs)
Department of Chemical Engineering, University of Arkansas, Fayetteville, AR 72701, USA
30秒速读
IN SHORT: This paper addresses the core challenge of accurately modeling context-dependent signaling, pathway cross-talk, and temporal dynamics across multiple biological scales in cellular signaling networks.
核心创新
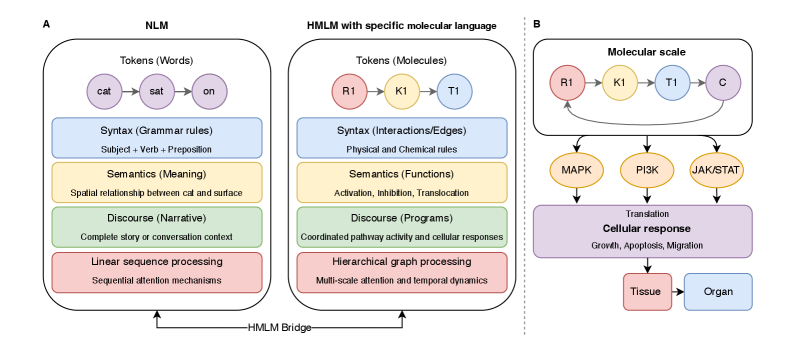
- Methodology Introduces cellular signaling as a molecular language with unique grammar and semantics, establishing a theoretical foundation for molecular artificial intelligence (MAI).
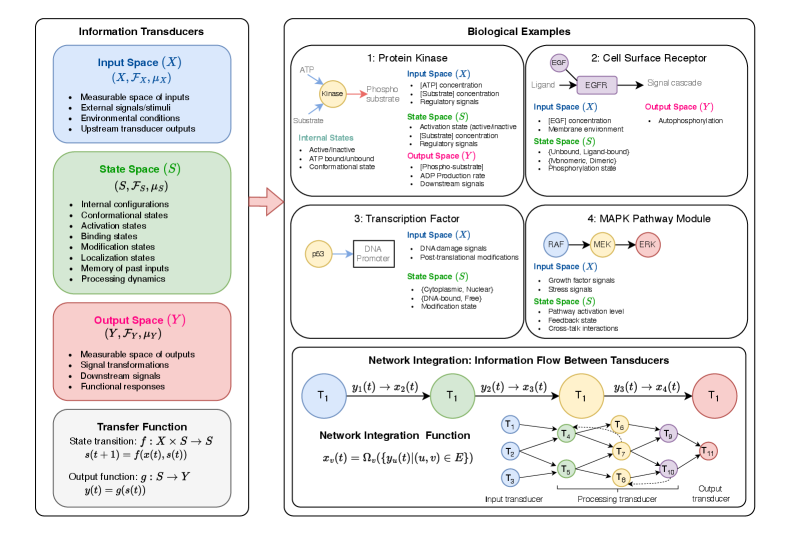
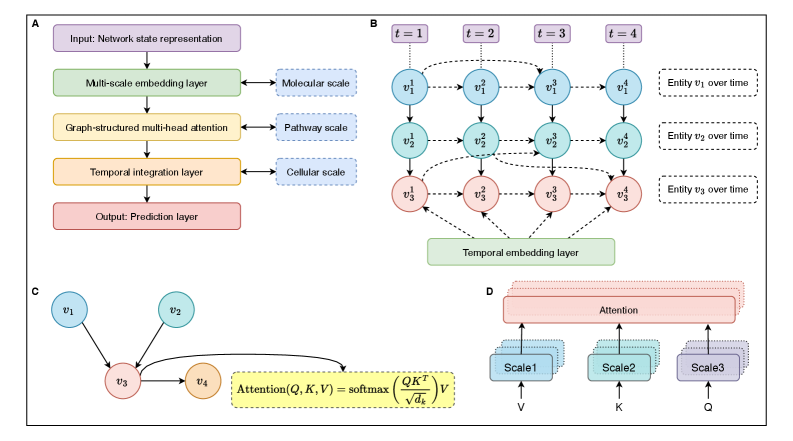
- Methodology Develops HMLMs as a novel computational architecture adapting transformer architecture to model signaling networks as information-processing systems across molecular, pathway, and cellular scales.
- Methodology Implements graph-structured attention mechanisms and hierarchical scale-bridging operators (aggregation, decomposition, translation) to accommodate signaling network topology and multi-scale organization.
主要结论
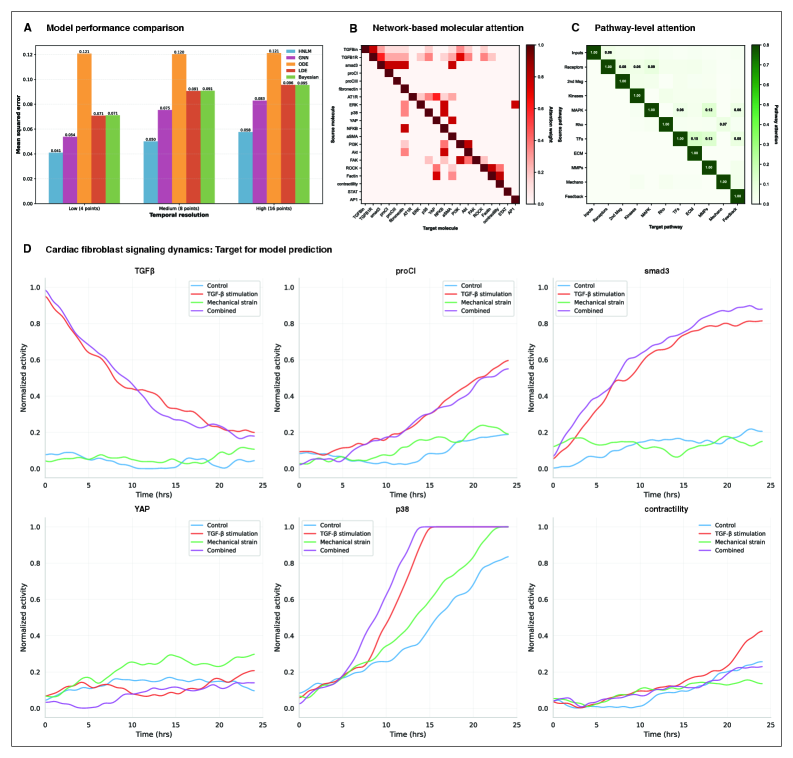
- HMLM achieved MSE of 0.058 for temporal signaling predictions, representing 30% improvement over GNNs (0.083) and 52% improvement over ODE models (0.121).
- Under sparse temporal sampling with only 4 timepoints, HMLM maintained superior performance with MSE = 0.041, demonstrating robustness to limited temporal data.
- Attention mechanisms identified biologically plausible pathway interactions including mechanotransduction-MAPK coupling and TGFβ to ERK signaling, validating the model's ability to capture meaningful biological relationships.
摘要: Cellular signaling networks represent complex information processing systems that have been modeled via traditional mathematical or statistical approaches. However, these methods often struggle to capture context-dependent signaling, pathway cross-talk, and temporal dynamics across multiple biological scales. Here, we introduce hierarchical molecular language models (HMLMs), a novel architecture that proposes a molecular network-specifiac large language model (LLM) to use in intracellular communication as a specialized molecular language, which includes molecules as tokens, protein interactions, post-translational modifications, and regulatory events modeled as semantic relationships within an adapted transformer architecture. HMLMs employ graph-structured attention mechanisms to accommodate signaling network topology while integrating information across the molecular, pathway, and cellular scales through hierarchical attention patterns. We demonstrate HMLM superiority using a cardiac fibroblast signaling network comprising over 100 molecular species across functional modules connected by regulatory edges. HMLM achieved a mean squared error (MSE) of 0.058 for temporal signaling predictions, representing 30% improvement over graph neural networks (GNNs: 0.083) and 52% improvement over ordinary differential equation models (ODEs: 0.121), with particular advantages under sparse temporal sampling conditions where HMLM maintained MSE = 0.041 with only 4 timepoints. The attention-based computational analysis identified key inter-pathway cross-talk patterns through learned attention mechanisms, including mechanotransduction-MAPK coupling and TGFβ to ERK signaling, demonstrating the model's capability to capture biologically plausible pathway interactions from network topology and temporal dynamics and convergent regulatory mechanisms controlling fibrosis markers in simulated cardiac fibroblast networks. The HMLMs offer a foundation for AI-driven biology and medicine with predictable scaling characteristics suitable for interactive applications. By bridging molecular mechanisms with cellular phenotypes through AI-driven molecular language representation, HMLMs provide a powerful paradigm for systems biology that advances precision medicine applications and therapeutic discovery in the era of AI.