
Paper List
-
Ill-Conditioning in Dictionary-Based Dynamic-Equation Learning: A Systems Biology Case Study
This paper addresses the critical challenge of numerical ill-conditioning and multicollinearity in library-based sparse regression methods (e.g., SIND...
-
Hybrid eTFCE–GRF: Exact Cluster-Size Retrieval with Analytical pp-Values for Voxel-Based Morphometry
This paper addresses the computational bottleneck in voxel-based neuroimaging analysis by providing a method that delivers exact cluster-size retrieva...
-
abx_amr_simulator: A simulation environment for antibiotic prescribing policy optimization under antimicrobial resistance
This paper addresses the critical challenge of quantitatively evaluating antibiotic prescribing policies under realistic uncertainty and partial obser...
-
PesTwin: a biology-informed Digital Twin for enabling precision farming
This paper addresses the critical bottleneck in precision agriculture: the inability to accurately forecast pest outbreaks in real-time, leading to su...
-
Equivariant Asynchronous Diffusion: An Adaptive Denoising Schedule for Accelerated Molecular Conformation Generation
This paper addresses the core challenge of generating physically plausible 3D molecular structures by bridging the gap between autoregressive methods ...
-
Omics Data Discovery Agents
This paper addresses the core challenge of making published omics data computationally reusable by automating the extraction, quantification, and inte...
-
Single-cell directional sensing at ultra-low chemoattractant concentrations from extreme first-passage events
This work addresses the core challenge of how a cell can rapidly and accurately determine the direction of a chemoattractant source when the signal is...
-
SDSR: A Spectral Divide-and-Conquer Approach for Species Tree Reconstruction
This paper addresses the computational bottleneck in reconstructing species trees from thousands of species and multiple genes by introducing a scalab...
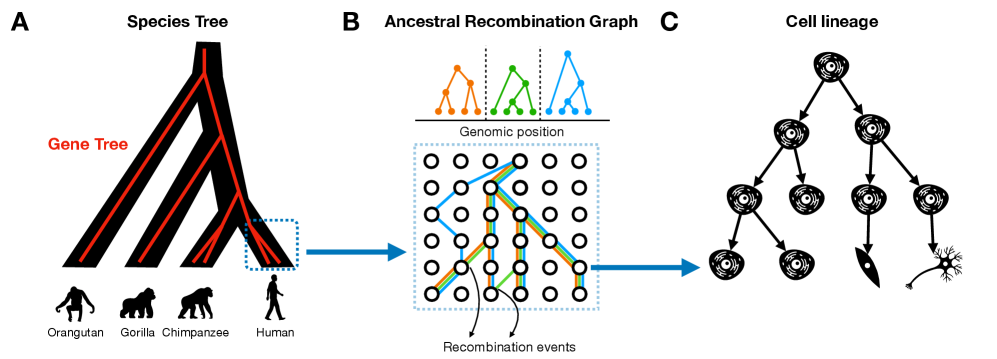
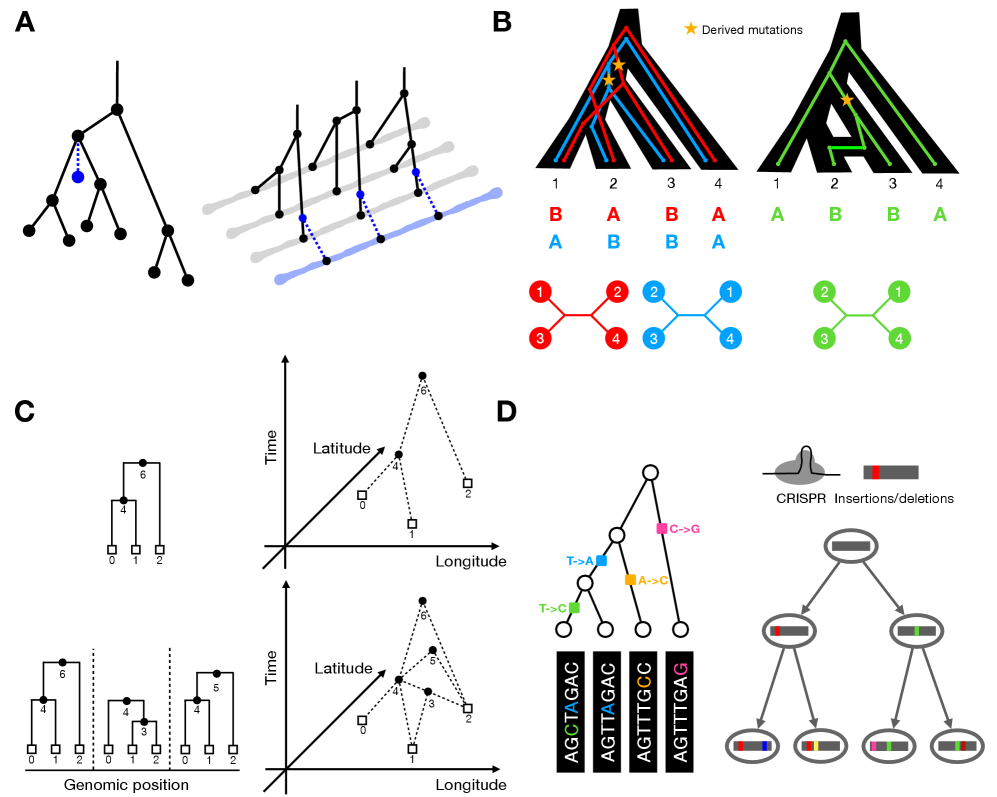
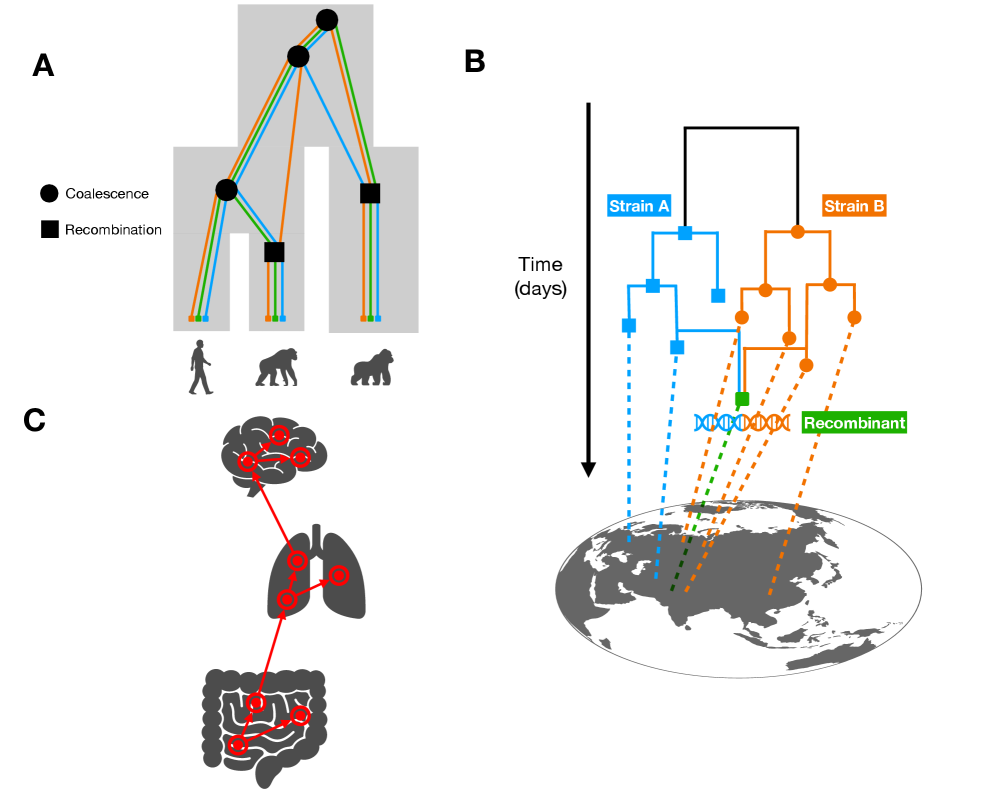
Tree Thinking in the Genomic Era: Unifying Models Across Cells, Populations, and Species
Stanford University | University of Oxford | University of California, Berkeley | Peking University | Guangzhou Medical University
30秒速读
IN SHORT: This paper addresses the fragmentation of tree-based inference methods across biological scales by identifying shared algorithmic principles and statistical challenges in phylogenetics, population genetics, and cell lineage tracing.
核心创新
- Methodology Identifies deep conceptual parallels between phylogenetic placement algorithms and ARG threading methods, demonstrating how phylogenetic placement generalizes to ARG reconstruction.
- Biology Shows that quartet-based network methods in phylogenetics and ABBA-BABA statistics in population genetics capture the same underlying signal of gene flow through asymmetric genealogical relationships.
- Methodology Demonstrates how ARG-based migration inference methods (e.g., GAIA, spacetrees) extend classical phylogeographic approaches by leveraging the full sequence of locally correlated genealogies along the genome.
主要结论
- Tree-based models provide a unified framework for ancestry inference across biological scales, with ARGs representing ~2.48 million SARS-CoV-2 genomes demonstrating pandemic-scale feasibility.
- Methodological parallels exist across domains: phylogenetic placement algorithms share core logic with ARG threading, and quartet-based methods in phylogenetics mirror ABBA-BABA statistics in population genetics for detecting gene flow.
- Current ARG inference algorithms remain constrained by simplifying assumptions (neutrality, panmixia, constant population size) and face challenges in uncertainty quantification, particularly for non-model species or limited sample sizes.
摘要: The ongoing explosion of genome sequence data is transforming how we reconstruct and understand the histories of biological systems. Across biological scales–from individual cells to populations and species–trees-based models provide a common framework for representing ancestry. Once limited to species phylogenetics, “tree thinking” now extends deeply to population genomics and cell biology, revealing the genealogical structure of genetic and phenotypic variation within and across organisms. Recently, there have been great methodological and computational advances on tree-based methods, including methods for inferring ancestral recombination graphs in populations, phylogenetic frameworks for comparative genomics, and lineage-tracing techniques in developmental and cancer biology. Despite differences in data types and biological contexts, these approaches share core statistical and algorithmic challenges: efficiently inferring branching histories from genomic information, integrating temporal and spatial signals, and connecting genealogical structures to evolutionary and functional processes. Recognizing these shared foundations opens opportunities for cross-fertilization between fields that are traditionally studied in isolation. By examining how tree-based methods are applied across cellular, population, and species scales, we identify the conceptual parallels that unite them and the distinct challenges that each domain presents. These comparisons offer new perspectives that can inform algorithmic innovations and lead to more powerful inference strategies across the full spectrum of biological systems.