
Paper List
-
Ill-Conditioning in Dictionary-Based Dynamic-Equation Learning: A Systems Biology Case Study
This paper addresses the critical challenge of numerical ill-conditioning and multicollinearity in library-based sparse regression methods (e.g., SIND...
-
Hybrid eTFCE–GRF: Exact Cluster-Size Retrieval with Analytical pp-Values for Voxel-Based Morphometry
This paper addresses the computational bottleneck in voxel-based neuroimaging analysis by providing a method that delivers exact cluster-size retrieva...
-
abx_amr_simulator: A simulation environment for antibiotic prescribing policy optimization under antimicrobial resistance
This paper addresses the critical challenge of quantitatively evaluating antibiotic prescribing policies under realistic uncertainty and partial obser...
-
PesTwin: a biology-informed Digital Twin for enabling precision farming
This paper addresses the critical bottleneck in precision agriculture: the inability to accurately forecast pest outbreaks in real-time, leading to su...
-
Equivariant Asynchronous Diffusion: An Adaptive Denoising Schedule for Accelerated Molecular Conformation Generation
This paper addresses the core challenge of generating physically plausible 3D molecular structures by bridging the gap between autoregressive methods ...
-
Omics Data Discovery Agents
This paper addresses the core challenge of making published omics data computationally reusable by automating the extraction, quantification, and inte...
-
Single-cell directional sensing at ultra-low chemoattractant concentrations from extreme first-passage events
This work addresses the core challenge of how a cell can rapidly and accurately determine the direction of a chemoattractant source when the signal is...
-
SDSR: A Spectral Divide-and-Conquer Approach for Species Tree Reconstruction
This paper addresses the computational bottleneck in reconstructing species trees from thousands of species and multiple genes by introducing a scalab...
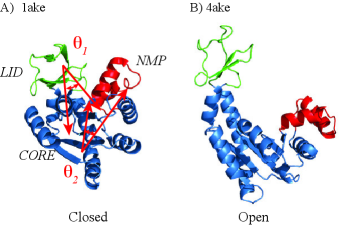
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
Department of Computer Science and Genome Center, University of California, Davis | Architecture et Dynamique des Macromolécules Biologiques, UMR 3528 du CNRS, Institut Pasteur | Department of Physics, School of Sciences, Great Bay University | Université Paris-Saclay, CNRS, CEA, Institut de Physique Théorique
30秒速读
IN SHORT: This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein conformations, a problem where existing methods often produce non-physical trajectories due to oversimplified energy surfaces and steric clashes.
核心创新
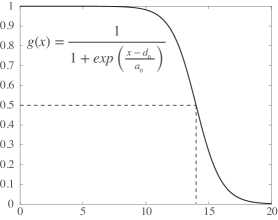
- Methodology Introduces SIDE (Stochastic Integro-Differential Equation), a novel Langevin bridge-based framework that efficiently approximates exact bridge equations at low temperatures to generate constrained transition trajectories.
- Methodology Develops a new coarse-grained potential that combines a Gō-like term (to preserve native backbone geometry) with a Rouse-type elastic energy term (from polymer physics), avoiding the problematic mixing of start/target conformation information used in prior methods like MinActionPath.
- Theory Provides a rigorous stochastic integro-differential formulation derived from the Langevin bridge formalism, which explicitly constrains trajectories to reach a target state within finite time, moving beyond Minimum Action Path (MAP) principles.
主要结论
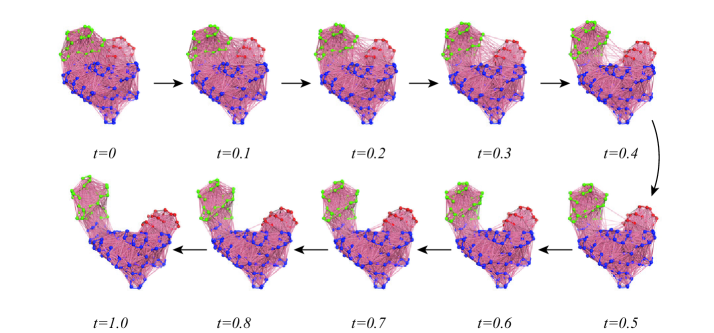
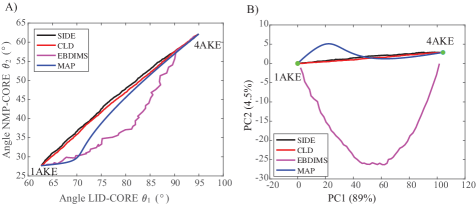
- The SIDE framework generates smooth, low-energy transition trajectories that maintain realistic molecular geometry, as demonstrated on several proteins undergoing large-scale conformational changes.
- SIDE frequently recovers experimentally supported intermediate states along transition paths, suggesting its paths have biological relevance beyond mere endpoint interpolation.
- Compared to established methods like MinActionPath and EBDIMS, SIDE offers improved physical realism and computational efficiency for modeling biomolecular conformational transitions, though challenges remain for highly complex motions.
摘要: We introduce a computational framework for generating realistic transition paths between distinct conformations of large biomolecular systems. The method is built on a stochastic integro-differential formulation derived from the Langevin bridge formalism, which constrains molecular trajectories to reach a prescribed final state within a finite time and yields an efficient low-temperature approximation of the exact bridge equation. To obtain physically meaningful protein transitions, we couple this formulation to a new coarse-grained potential combining a Gō-like term that preserves native backbone geometry with a Rouse-type elastic energy term from polymer physics; we refer to the resulting approach as SIDE. We evaluate SIDE on several proteins undergoing large-scale conformational changes and compare its performance with established methods such as MinActionPath and EBDIMS. SIDE generates smooth, low-energy trajectories that maintain molecular geometry and frequently recover experimentally supported intermediate states. Although challenges remain for highly complex motions—largely due to the simplified coarse-grained potential—our results demonstrate that SIDE offers a powerful and computationally efficient strategy for modeling biomolecular conformational transitions.