
Paper List
-
Autonomous Agents Coordinating Distributed Discovery Through Emergent Artifact Exchange
This paper addresses the fundamental limitation of current AI-assisted scientific research by enabling truly autonomous, decentralized investigation w...
-
D-MEM: Dopamine-Gated Agentic Memory via Reward Prediction Error Routing
This paper addresses the fundamental scalability bottleneck in LLM agentic memory systems: the O(N²) computational complexity and unbounded API token ...
-
Countershading coloration in blue shark skin emerges from hierarchically organized and spatially tuned photonic architectures inside skin denticles
This paper solves the core problem of how blue sharks achieve their striking dorsoventral countershading camouflage, revealing that coloration origina...
-
Human-like Object Grouping in Self-supervised Vision Transformers
This paper addresses the core challenge of quantifying how well self-supervised vision models capture human-like object grouping in natural scenes, br...
-
Hierarchical pp-Adic Framework for Gene Regulatory Networks: Theory and Stability Analysis
This paper addresses the core challenge of mathematically capturing the inherent hierarchical organization and multi-scale stability of gene regulator...
-
Towards unified brain-to-text decoding across speech production and perception
This paper addresses the core challenge of developing a unified brain-to-text decoding framework that works across both speech production and percepti...
-
Dual-Laws Model for a theory of artificial consciousness
This paper addresses the core challenge of developing a comprehensive, testable theory of consciousness that bridges biological and artificial systems...
-
Pulse desynchronization of neural populations by targeting the centroid of the limit cycle in phase space
This work addresses the core challenge of determining optimal pulse timing and intensity for desynchronizing pathological neural oscillations when the...
Tree Thinking in the Genomic Era: Unifying Models Across Cells, Populations, and Species
Stanford University | University of Oxford | University of California, Berkeley | Peking University | Guangzhou Medical University
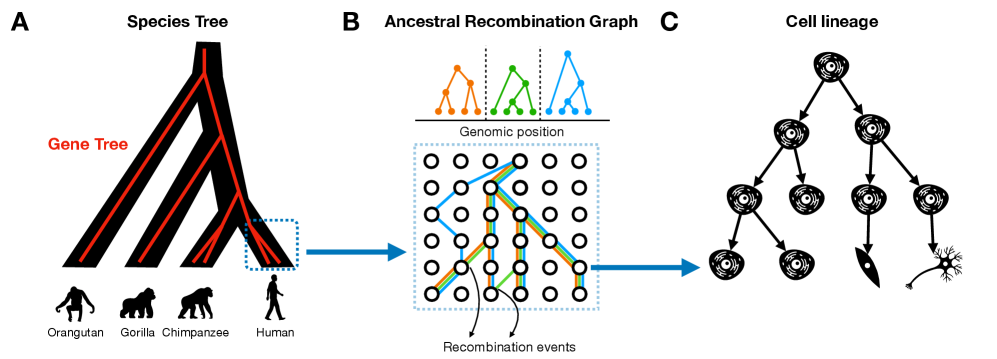
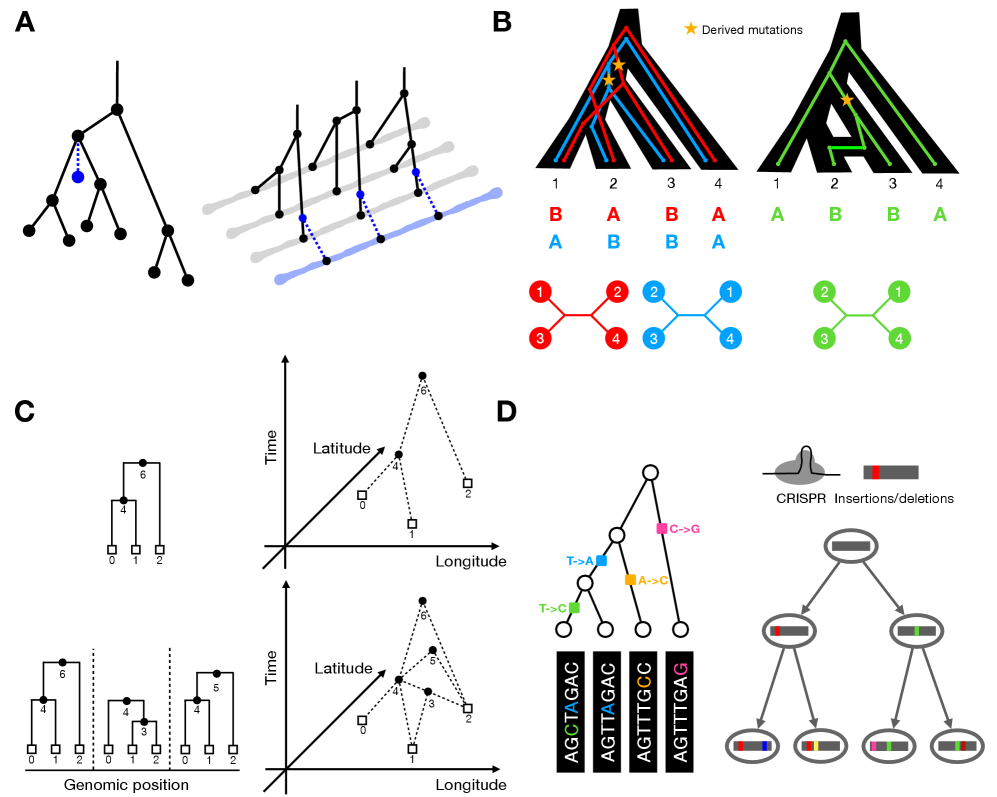
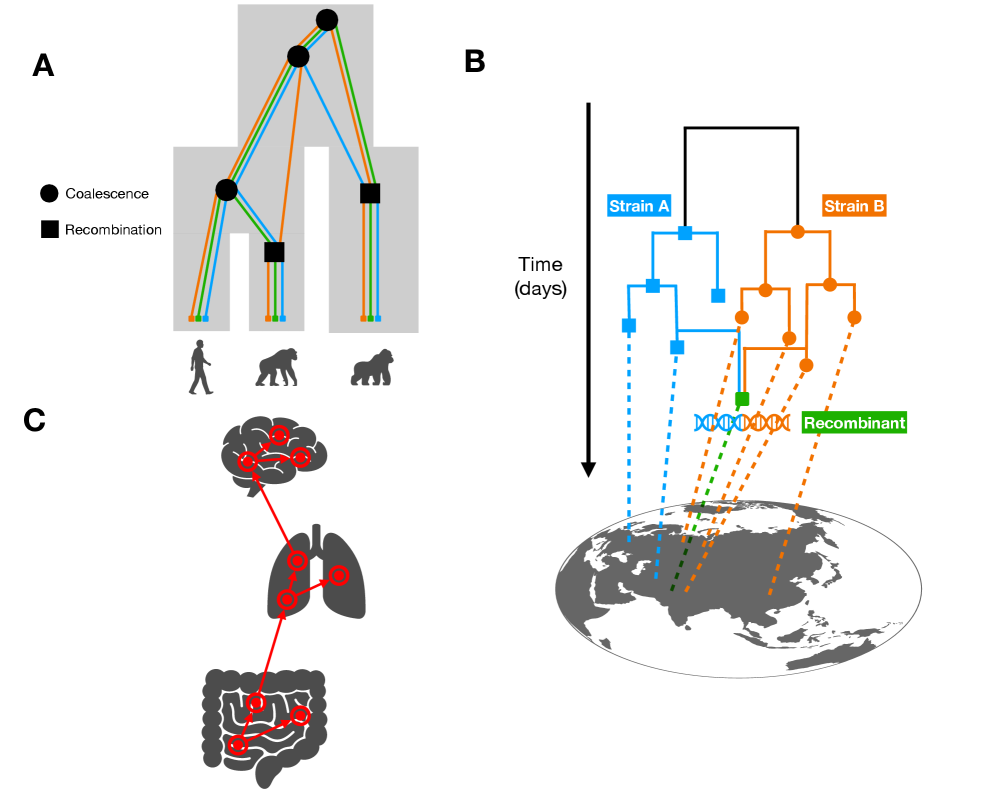
30秒速读
IN SHORT: This paper addresses the fragmentation of tree-based inference methods across biological scales by identifying shared algorithmic principles and statistical challenges in phylogenetics, population genetics, and cell lineage tracing.
核心创新
- Methodology Identifies deep conceptual parallels between phylogenetic placement algorithms and ARG threading methods, demonstrating how phylogenetic placement generalizes to ARG reconstruction.
- Biology Shows that quartet-based network methods in phylogenetics and ABBA-BABA statistics in population genetics capture the same underlying signal of gene flow through asymmetric genealogical relationships.
- Methodology Demonstrates how ARG-based migration inference methods (e.g., GAIA, spacetrees) extend classical phylogeographic approaches by leveraging the full sequence of locally correlated genealogies along the genome.
主要结论
- Tree-based models provide a unified framework for ancestry inference across biological scales, with ARGs representing ~2.48 million SARS-CoV-2 genomes demonstrating pandemic-scale feasibility.
- Methodological parallels exist across domains: phylogenetic placement algorithms share core logic with ARG threading, and quartet-based methods in phylogenetics mirror ABBA-BABA statistics in population genetics for detecting gene flow.
- Current ARG inference algorithms remain constrained by simplifying assumptions (neutrality, panmixia, constant population size) and face challenges in uncertainty quantification, particularly for non-model species or limited sample sizes.
摘要: The ongoing explosion of genome sequence data is transforming how we reconstruct and understand the histories of biological systems. Across biological scales–from individual cells to populations and species–trees-based models provide a common framework for representing ancestry. Once limited to species phylogenetics, “tree thinking” now extends deeply to population genomics and cell biology, revealing the genealogical structure of genetic and phenotypic variation within and across organisms. Recently, there have been great methodological and computational advances on tree-based methods, including methods for inferring ancestral recombination graphs in populations, phylogenetic frameworks for comparative genomics, and lineage-tracing techniques in developmental and cancer biology. Despite differences in data types and biological contexts, these approaches share core statistical and algorithmic challenges: efficiently inferring branching histories from genomic information, integrating temporal and spatial signals, and connecting genealogical structures to evolutionary and functional processes. Recognizing these shared foundations opens opportunities for cross-fertilization between fields that are traditionally studied in isolation. By examining how tree-based methods are applied across cellular, population, and species scales, we identify the conceptual parallels that unite them and the distinct challenges that each domain presents. These comparisons offer new perspectives that can inform algorithmic innovations and lead to more powerful inference strategies across the full spectrum of biological systems.