
Paper List
-
Autonomous Agents Coordinating Distributed Discovery Through Emergent Artifact Exchange
This paper addresses the fundamental limitation of current AI-assisted scientific research by enabling truly autonomous, decentralized investigation w...
-
D-MEM: Dopamine-Gated Agentic Memory via Reward Prediction Error Routing
This paper addresses the fundamental scalability bottleneck in LLM agentic memory systems: the O(N²) computational complexity and unbounded API token ...
-
Countershading coloration in blue shark skin emerges from hierarchically organized and spatially tuned photonic architectures inside skin denticles
This paper solves the core problem of how blue sharks achieve their striking dorsoventral countershading camouflage, revealing that coloration origina...
-
Human-like Object Grouping in Self-supervised Vision Transformers
This paper addresses the core challenge of quantifying how well self-supervised vision models capture human-like object grouping in natural scenes, br...
-
Hierarchical pp-Adic Framework for Gene Regulatory Networks: Theory and Stability Analysis
This paper addresses the core challenge of mathematically capturing the inherent hierarchical organization and multi-scale stability of gene regulator...
-
Towards unified brain-to-text decoding across speech production and perception
This paper addresses the core challenge of developing a unified brain-to-text decoding framework that works across both speech production and percepti...
-
Dual-Laws Model for a theory of artificial consciousness
This paper addresses the core challenge of developing a comprehensive, testable theory of consciousness that bridges biological and artificial systems...
-
Pulse desynchronization of neural populations by targeting the centroid of the limit cycle in phase space
This work addresses the core challenge of determining optimal pulse timing and intensity for desynchronizing pathological neural oscillations when the...
Unlocking hidden biomolecular conformational landscapes in diffusion models at inference time
Stanford University | Yale School of Medicine

30秒速读
IN SHORT: This paper addresses the core challenge of efficiently and accurately sampling the conformational landscape of biomolecules from diffusion-based structure prediction models, which typically output highly concentrated distributions around a single static structure.
核心创新
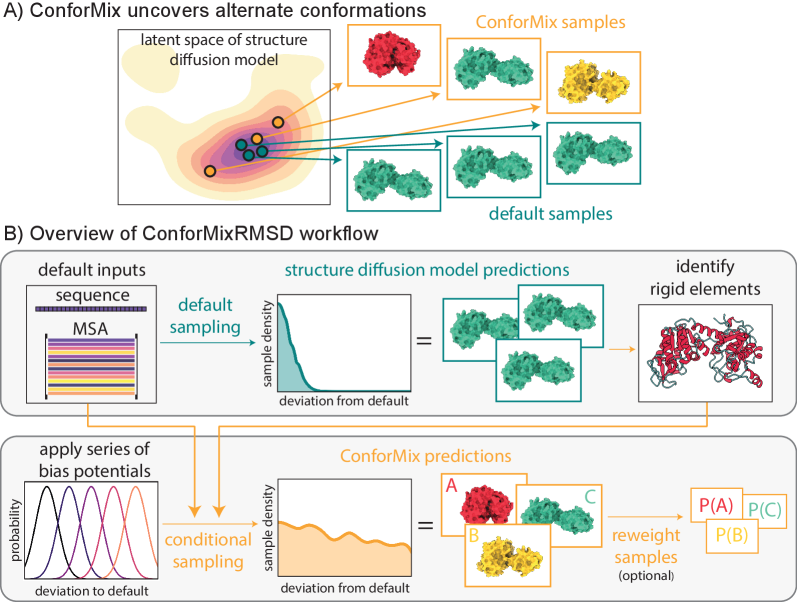
- Methodology Introduces ConforMix, a novel inference-time algorithm combining twisted sequential Monte Carlo (SMC) with automated exploration of the diffusion landscape, enabling asymptotically exact sampling of conditional distributions without additional model training.
- Methodology Presents ConforMixRMSD, an instantiation for automated exploration that biases sampling away from the default prediction using RMSD-based potentials on rigid secondary structure elements, recovering diverse conformations without prior knowledge of degrees of freedom.
- Methodology Applies the multistate Bennett acceptance ratio (MBAR) free energy estimation algorithm to diffusion models for the first time, enabling reconstruction of the unbiased model landscape from conditional samples.
主要结论
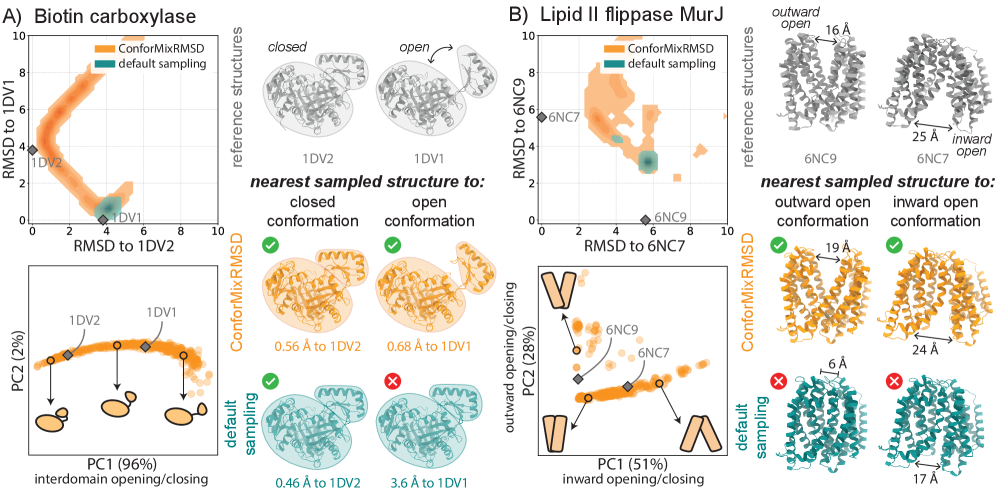
- ConforMixRMSD applied to Boltz-1 (an AlphaFold 3-like model) significantly outperforms MSA-modification baselines (AFCluster, AFSample2, CF-random) in recovering experimentally observed alternative conformations for domain motion (coverage: 0.69 ± 0.15 vs. 0.51 ± 0.17 for best baseline), membrane transporter (0.33 ± 0.23 vs. 0.20 ± 0.20), and cryptic pocket (0.45 ± 0.18 vs. 0.39 ± 0.16) protein sets, as measured by coverage at 50% of reference-to-reference RMSD.
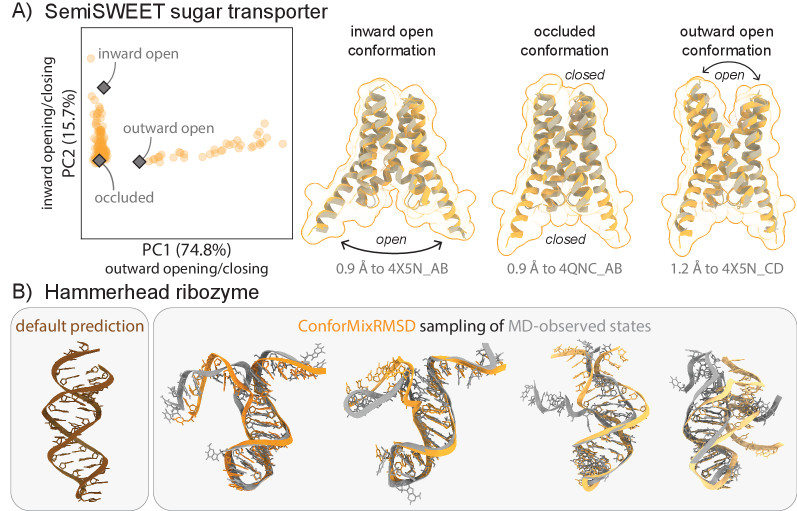
- The method captures biologically relevant conformational transitions (domain motion, transporter cycling, cryptic pocket flexibility) while avoiding unphysical states through filtering based on pLDDT values and clash detection, demonstrating its utility for exploring continuous transitions.
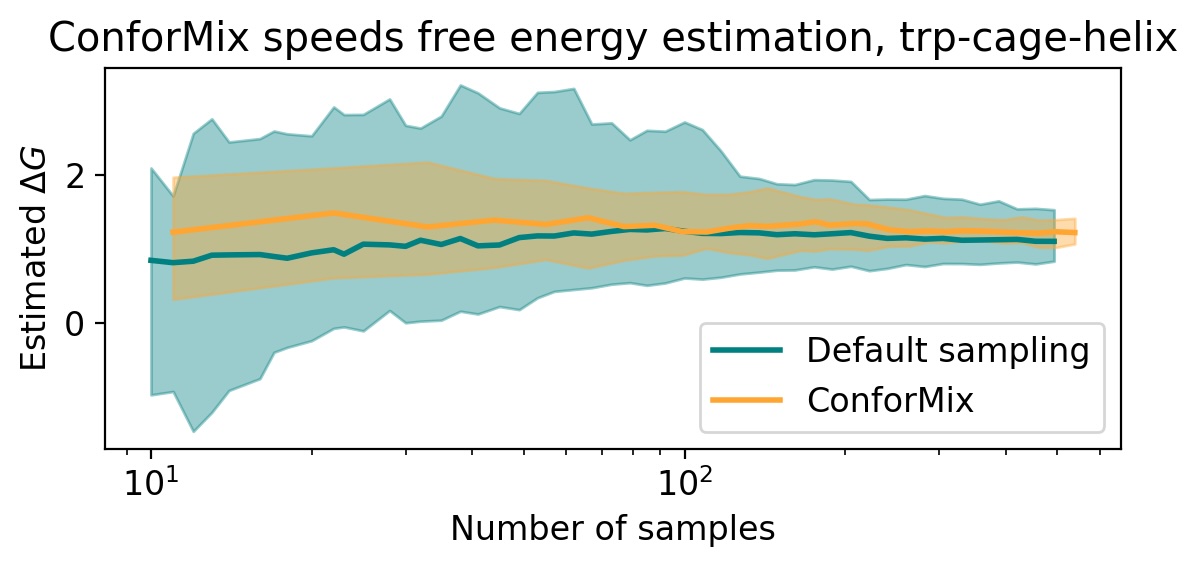
- ConforMix enables efficient free energy estimation when applied to models like BioEmu, boosting the speed of such calculations, and its framework is orthogonal to model pretraining improvements, meaning it would benefit even a hypothetical model that perfectly reproduces the Boltzmann distribution.
摘要: The function of biomolecules such as proteins depends on their ability to interconvert between a wide range of structures or “conformations.” Researchers have endeavored for decades to develop computational methods to predict the distribution of conformations, which is far harder to determine experimentally than a static folded structure. We present ConforMix, an inference-time algorithm that enhances sampling of conformational distributions using a combination of classifier guidance, filtering, and free energy estimation. Our approach upgrades diffusion models—whether trained for static structure prediction or conformational generation—to enable more efficient discovery of conformational variability without requiring prior knowledge of major degrees of freedom. ConforMix is orthogonal to improvements in model pretraining and would benefit even a hypothetical model that perfectly reproduced the Boltzmann distribution. Remarkably, when applied to a diffusion model trained for static structure prediction, ConforMix captures structural changes including domain motion, cryptic pocket flexibility, and transporter cycling, while avoiding unphysical states. Case studies of biologically critical proteins demonstrate the scalability, accuracy, and utility of this method.