
Paper List
-
Developing the PsyCogMetrics™ AI Lab to Evaluate Large Language Models and Advance Cognitive Science
This paper addresses the critical gap between sophisticated LLM evaluation needs and the lack of accessible, scientifically rigorous platforms that in...
-
Equivalence of approximation by networks of single- and multi-spike neurons
This paper resolves the fundamental question of whether single-spike spiking neural networks (SNNs) are inherently less expressive than multi-spike SN...
-
The neuroscience of transformers
提出了Transformer架构与皮层柱微环路之间的新颖计算映射,连接了现代AI与神经科学。
-
Framing local structural identifiability and observability in terms of parameter-state symmetries
This paper addresses the core challenge of systematically determining which parameters and states in a mechanistic ODE model can be uniquely inferred ...
-
Leveraging Phytolith Research using Artificial Intelligence
This paper addresses the critical bottleneck in phytolith research by automating the labor-intensive manual microscopy process through a multimodal AI...
-
Neural network-based encoding in free-viewing fMRI with gaze-aware models
This paper addresses the core challenge of building computationally efficient and ecologically valid brain encoding models for naturalistic vision by ...
-
Scalable DNA Ternary Full Adder Enabled by a Competitive Blocking Circuit
This paper addresses the core bottleneck of carry information attenuation and limited computational scale in DNA binary adders by introducing a scalab...
-
ELISA: An Interpretable Hybrid Generative AI Agent for Expression-Grounded Discovery in Single-Cell Genomics
This paper addresses the critical bottleneck of translating high-dimensional single-cell transcriptomic data into interpretable biological hypotheses ...
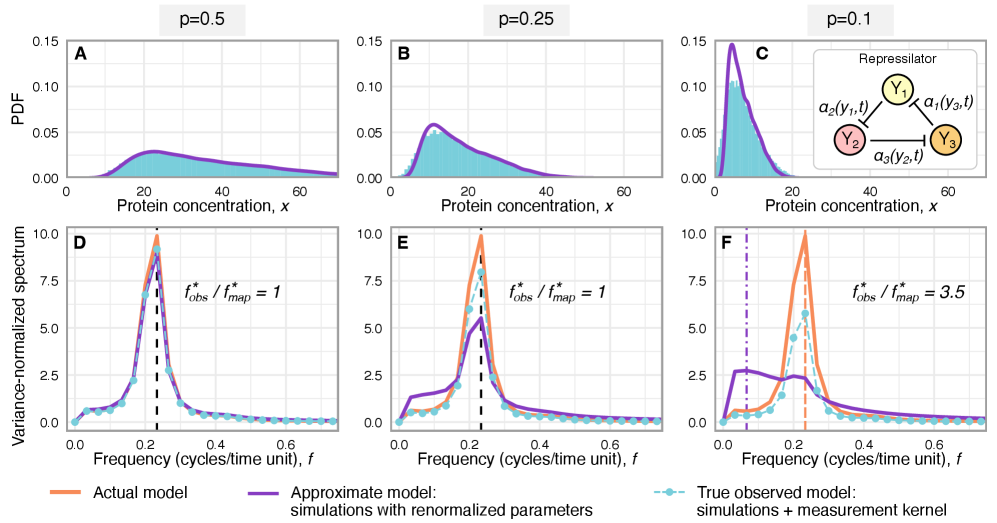
Imperfect molecular detection renormalizes apparent kinetic rates in stochastic gene regulatory networks
Department of Mathematical Analysis and Numerical Mathematics, Comenius University, Slovakia | University of Edinburgh, UK
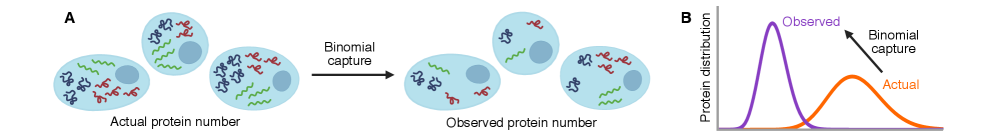
30秒速读
IN SHORT: This paper addresses the core challenge of distinguishing genuine stochastic dynamics of gene regulatory networks from artifacts introduced by imperfect molecular detection in single-cell experiments.
核心创新
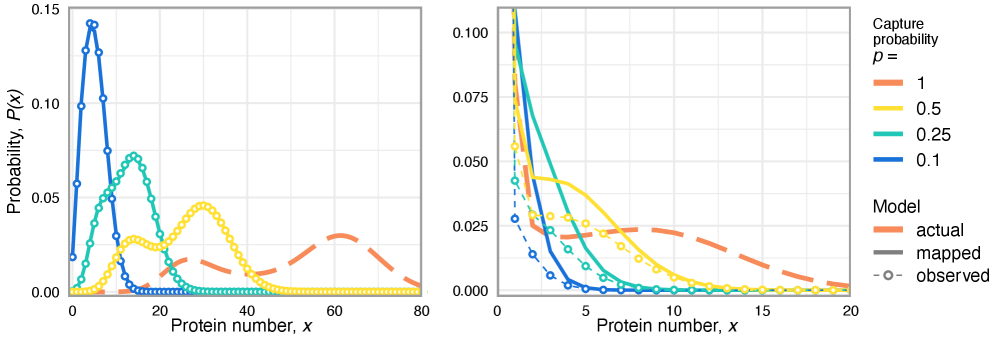
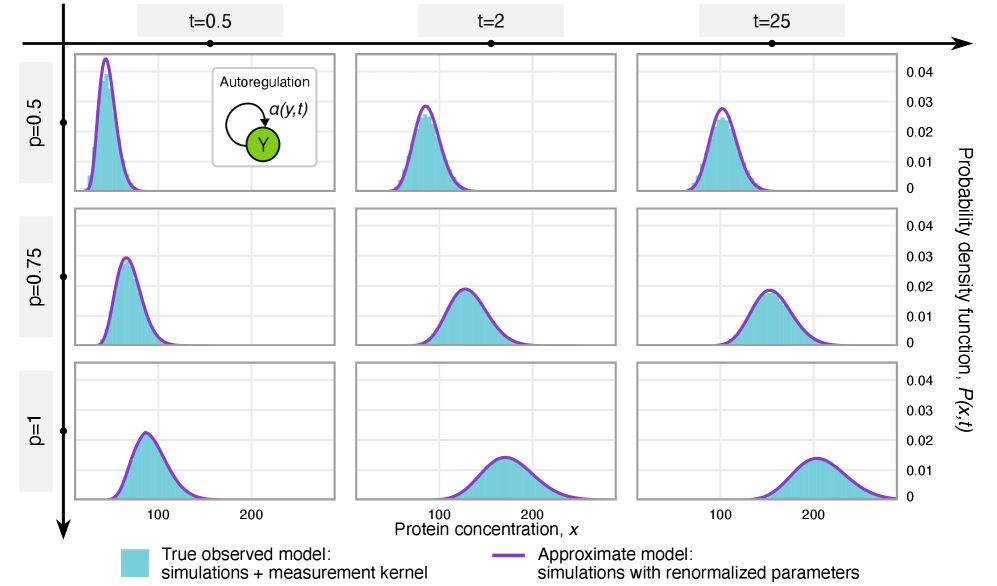
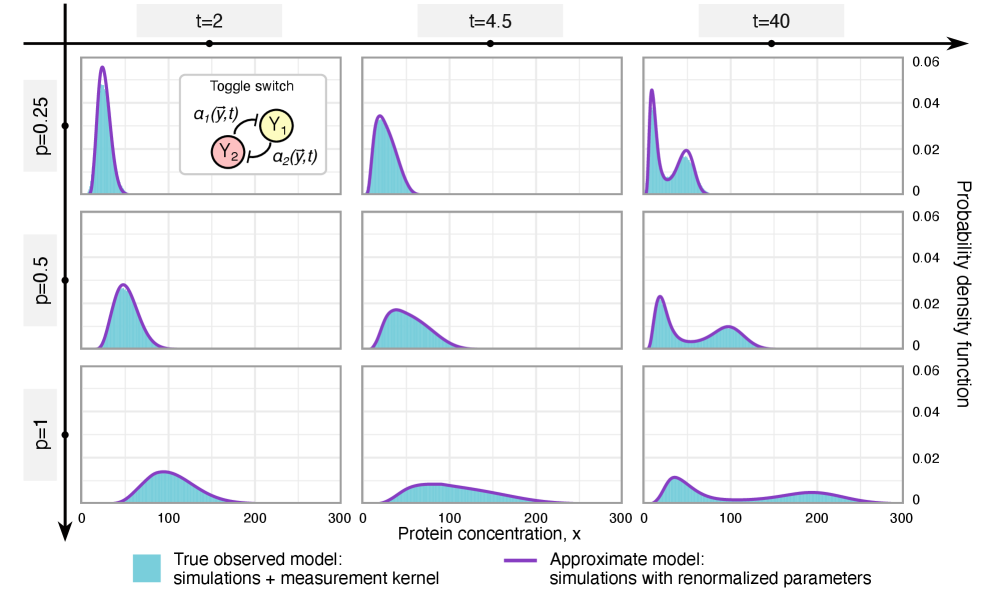
- Methodology Extends the binomial capture model from simple gene expression to general gene regulatory networks (GRNs) with explicit regulation, enabling analysis of technical noise in complex systems.
- Theory Establishes precise mathematical conditions under which technical noise leads to a renormalization (rescaling) of kinetic rates versus when it introduces non-absorbable distortions.
- Methodology Derives results valid for networks of arbitrary connectivity and under time-dependent kinetic rates, significantly generalizing previous steady-state analyses.
主要结论
- Technical noise systematically reduces the apparent mean burst size of gene products by a factor of p (the capture probability), e.g., from b(t) to b(t)*p.
- Rate renormalization occurs when promoter-state transitions are on a distinct timescale (much slower/faster) than other reactions or under high transcription factor abundance.
- The framework shows that for the telegraph model, the observed mRNA dynamics are equivalent to the true system with a renormalized transcription rate: k₃(t) → p*k₃(t).
摘要: Imperfect molecular detection in single-cell experiments introduces technical noise that obscures the true stochastic dynamics of gene regulatory networks. While binomial models of molecular capture provide a principled description of imperfect detection, they have so far been analyzed only for simple gene-expression models that do not explicitly account for regulation. Here, we extend binomial models of capture to general gene regulatory networks to understand how imperfect capture reshapes the observed time-dependent statistics of molecular counts. Our results reveal when capture effects correspond to a renormalization of a subset of the kinetic rates and when they cannot be absorbed into effective rates, providing a systematic basis for interpreting noisy single-cell measurements. In particular, we show that rate renormalization emerges either under significant transcription factor abundance or when promoter-state transitions occur on a distinct (much slower or faster) timescale than other reactions. In these cases, technical noise causes the apparent mean burst size of synthesized gene products to appear reduced while transcription factor binding reactions appear faster. These effects hold for gene regulatory networks of arbitrary connectivity and remain valid under time-dependent kinetic rates.