
Paper List
-
Developing the PsyCogMetrics™ AI Lab to Evaluate Large Language Models and Advance Cognitive Science
This paper addresses the critical gap between sophisticated LLM evaluation needs and the lack of accessible, scientifically rigorous platforms that in...
-
Equivalence of approximation by networks of single- and multi-spike neurons
This paper resolves the fundamental question of whether single-spike spiking neural networks (SNNs) are inherently less expressive than multi-spike SN...
-
The neuroscience of transformers
提出了Transformer架构与皮层柱微环路之间的新颖计算映射,连接了现代AI与神经科学。
-
Framing local structural identifiability and observability in terms of parameter-state symmetries
This paper addresses the core challenge of systematically determining which parameters and states in a mechanistic ODE model can be uniquely inferred ...
-
Leveraging Phytolith Research using Artificial Intelligence
This paper addresses the critical bottleneck in phytolith research by automating the labor-intensive manual microscopy process through a multimodal AI...
-
Neural network-based encoding in free-viewing fMRI with gaze-aware models
This paper addresses the core challenge of building computationally efficient and ecologically valid brain encoding models for naturalistic vision by ...
-
Scalable DNA Ternary Full Adder Enabled by a Competitive Blocking Circuit
This paper addresses the core bottleneck of carry information attenuation and limited computational scale in DNA binary adders by introducing a scalab...
-
ELISA: An Interpretable Hybrid Generative AI Agent for Expression-Grounded Discovery in Single-Cell Genomics
This paper addresses the critical bottleneck of translating high-dimensional single-cell transcriptomic data into interpretable biological hypotheses ...
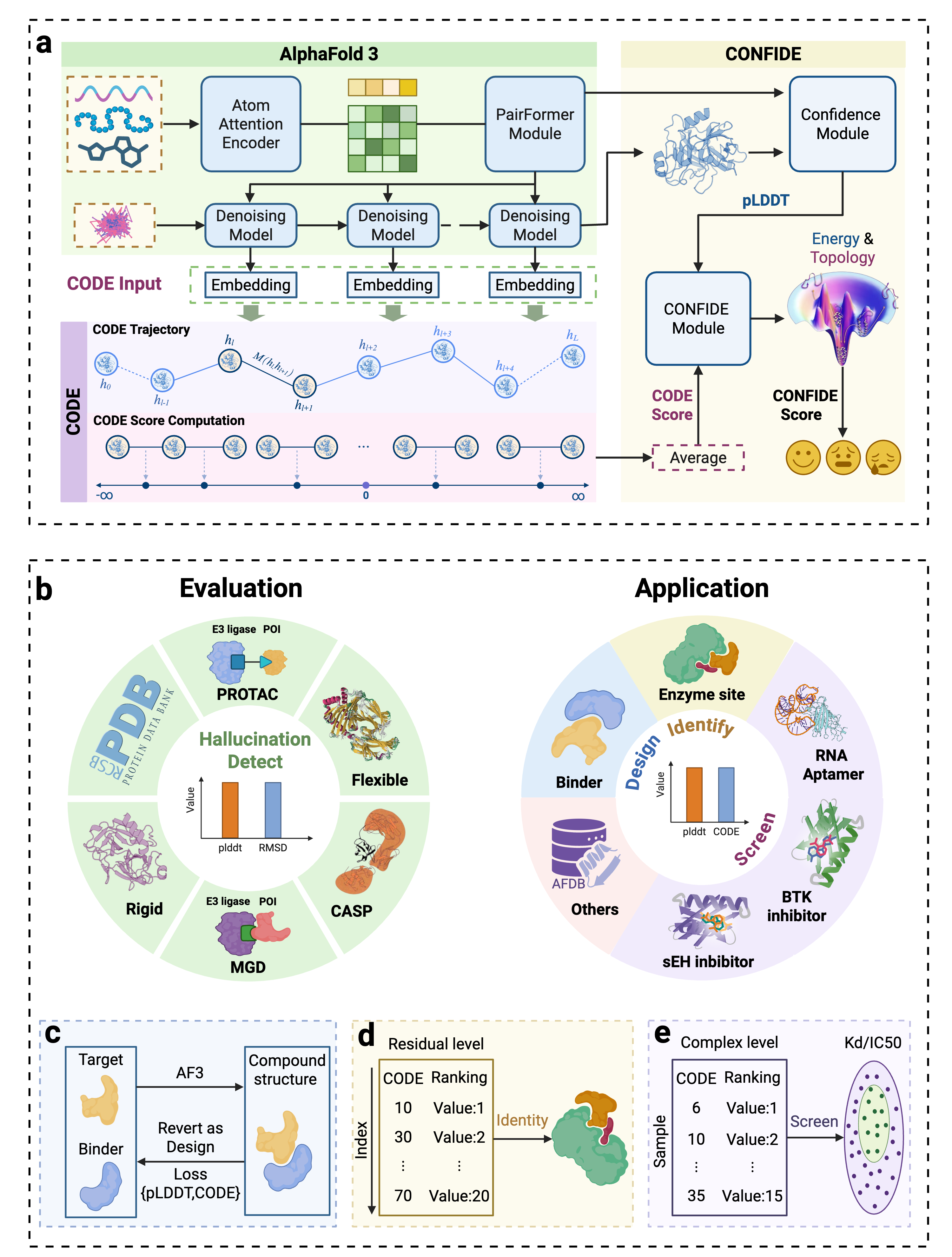
CONFIDE: Hallucination Assessment for Reliable Biomolecular Structure Prediction and Design
The Chinese University of Hong Kong | Zhejiang University | Macao Polytechnic University | University of Electronic Science and Technology of China
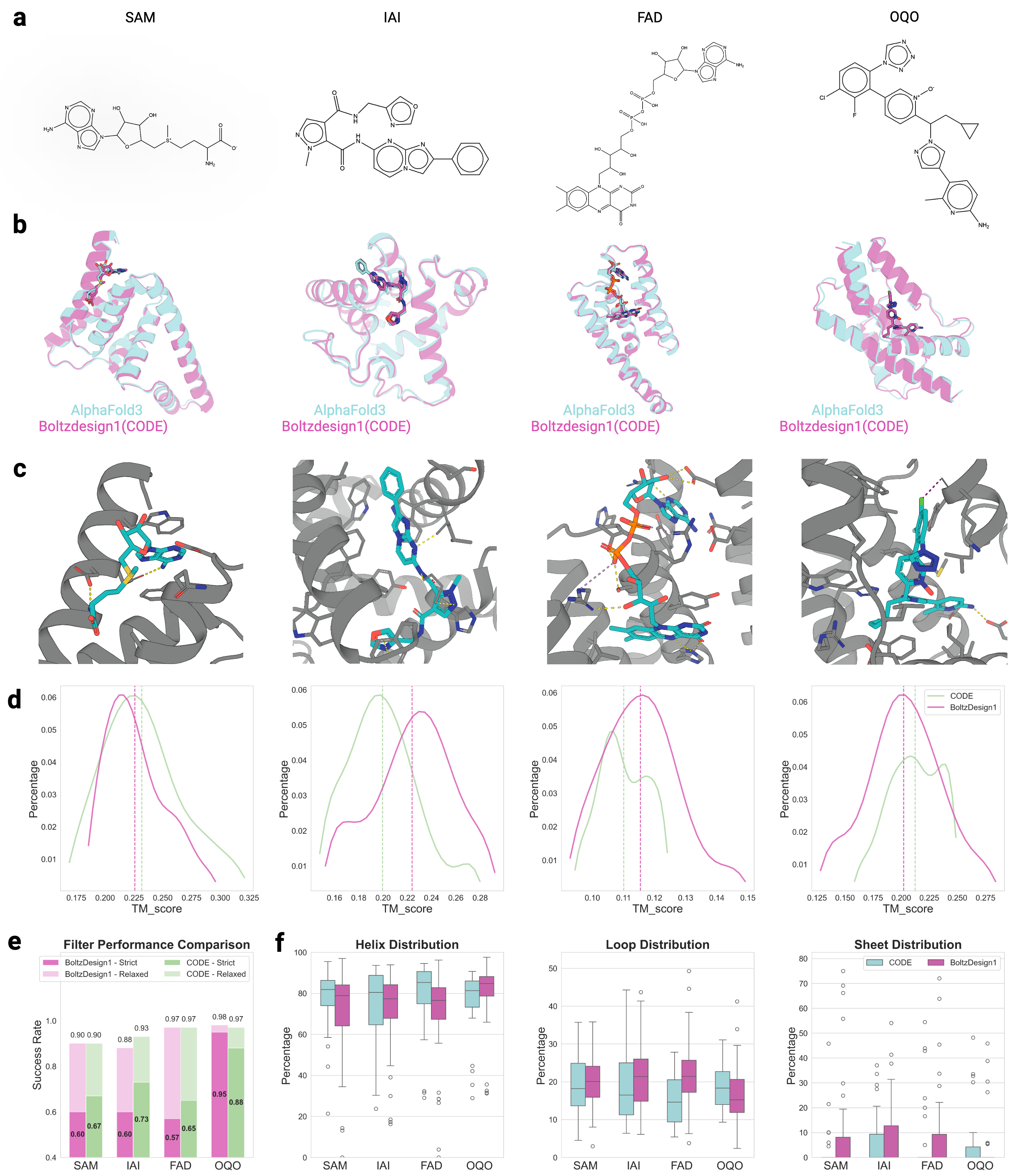
30秒速读
IN SHORT: This paper addresses the critical limitation of current protein structure prediction models (like AlphaFold3) where high-confidence scores (pLDDT) can be misleading, failing to detect subtle structural errors like atomic clashes and topological traps, which undermines reliability in downstream applications like drug discovery.
核心创新
- Methodology Introduces CODE (Chain of Diffusion Embeddings), a novel, unsupervised metric derived from AlphaFold3's latent diffusion embeddings that directly quantifies topological frustration, a key factor in protein folding kinetics previously overlooked by confidence scores.
- Methodology Proposes CONFIDE, a unified evaluation framework that integrates the energetic perspective of pLDDT with the topological perspective of CODE, providing a more comprehensive and reliable assessment of predicted biomolecular structures.
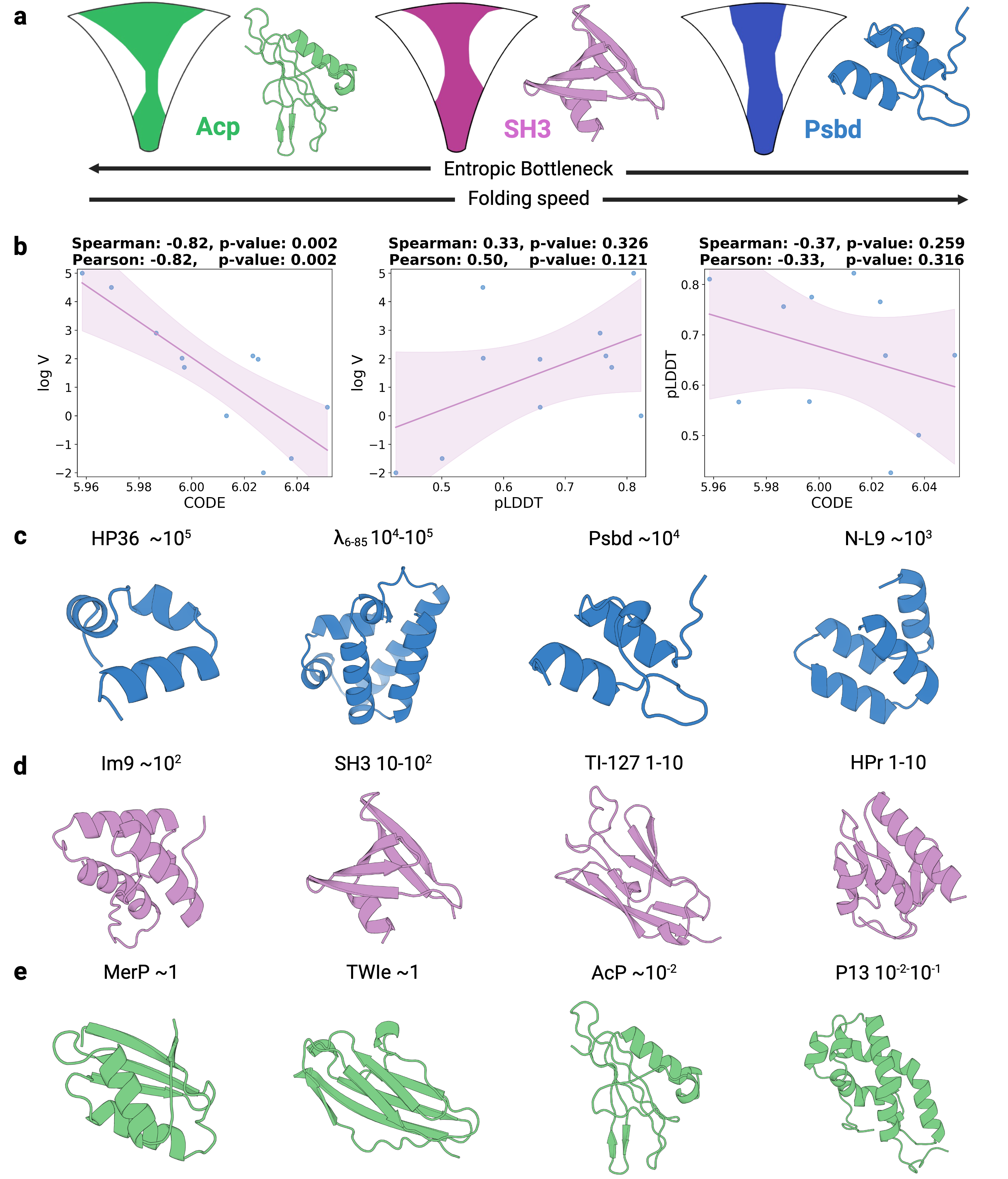
- Biology Establishes a strong empirical link between the CODE metric and protein folding rates driven by topological frustration (Spearman correlation of -0.82, p=0.002), offering a data-driven proxy for a complex biophysical phenomenon.
主要结论
- CODE demonstrates a strong, statistically significant correlation with protein folding rates mediated by topological frustration (Spearman ρ = -0.82, p=0.002), far outperforming pLDDT (ρ = 0.33, p=0.326).
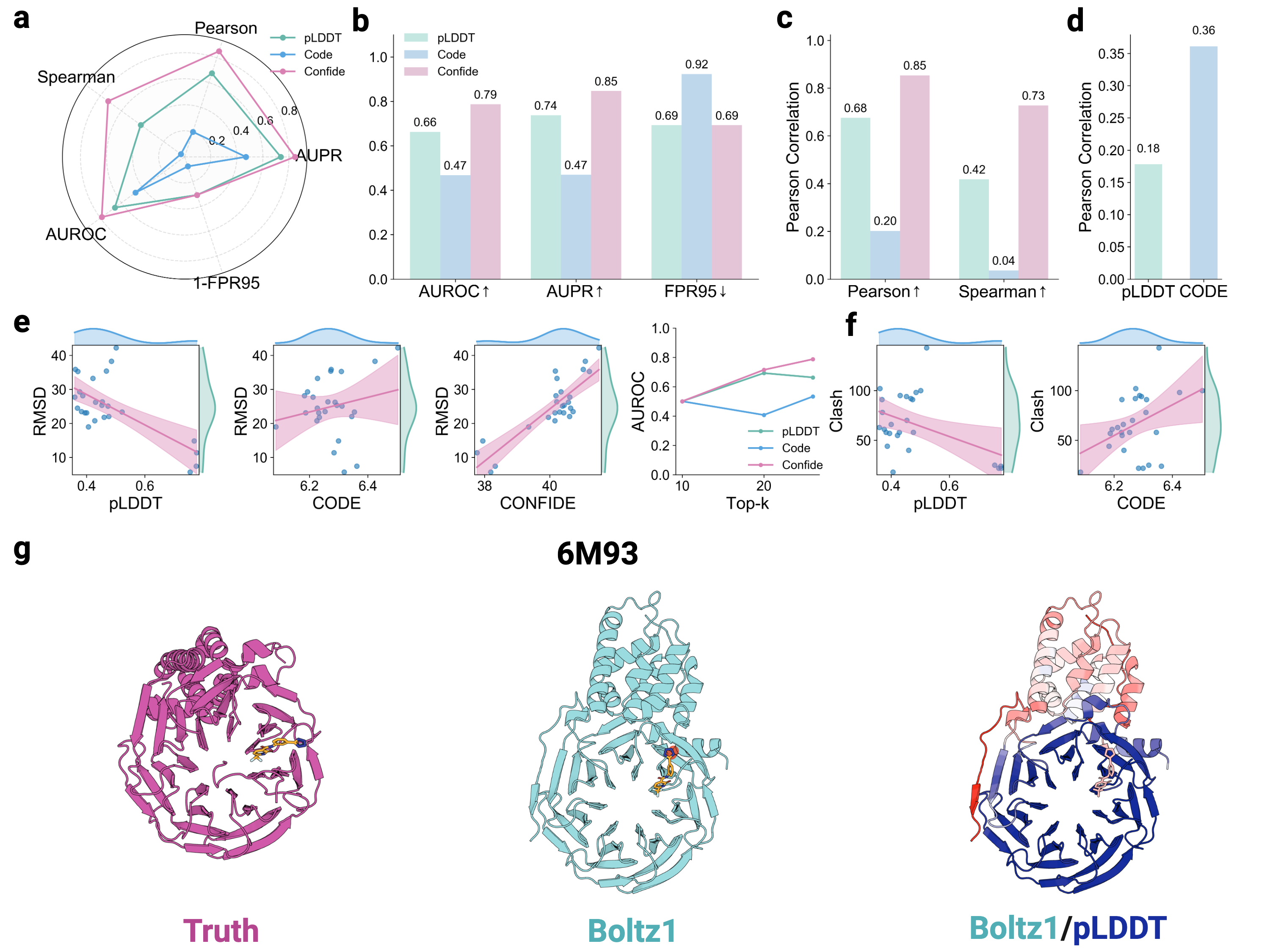
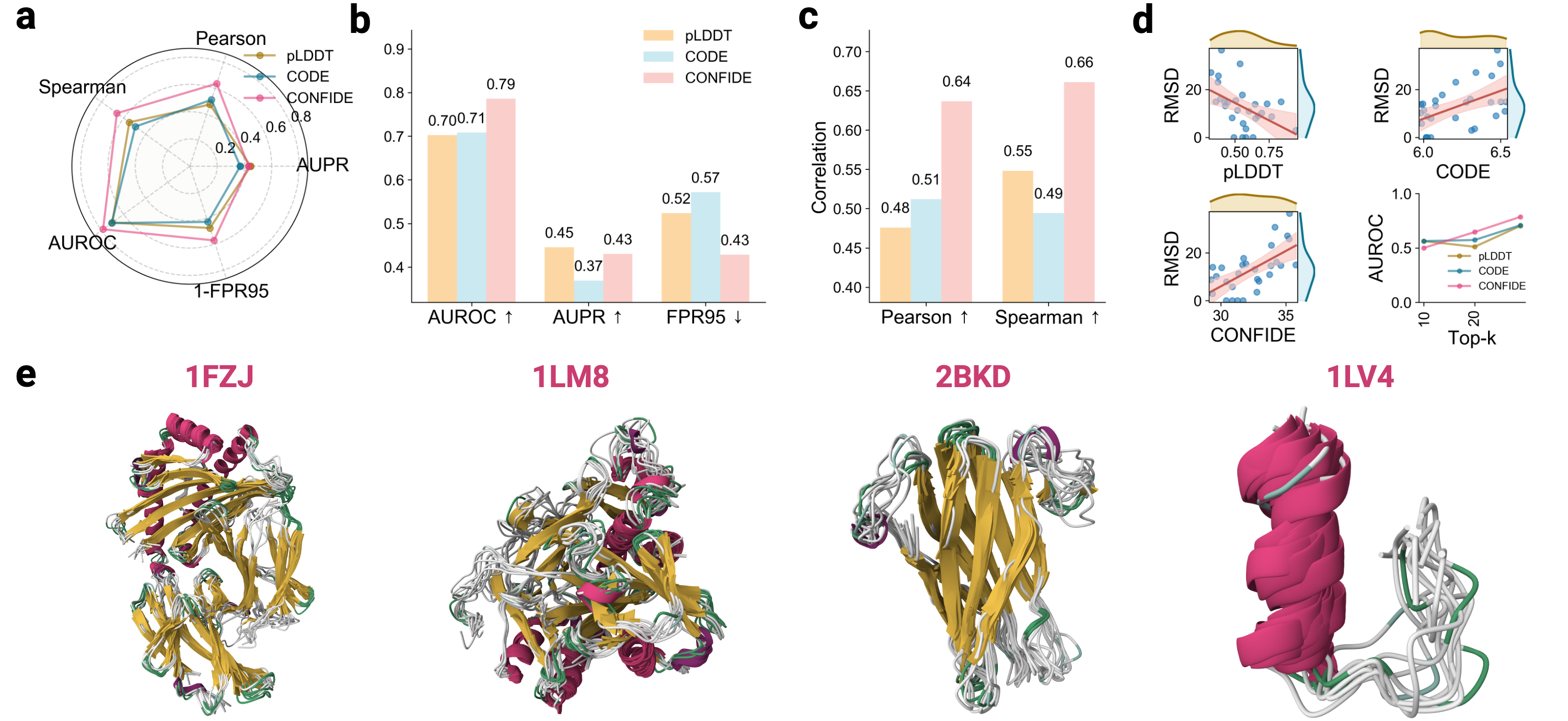
- The CONFIDE framework significantly improves hallucination detection, achieving a Spearman correlation of 0.73 with RMSD on molecular glue benchmarks, a 73.8% relative improvement over pLDDT's correlation of 0.42.
- CONFIDE enables practical downstream applications, improving binder design success rates (e.g., +13% for IAI) and accurately predicting mutation-induced binding affinity changes (Spearman ρ = 0.83 for BTK vs. Fenebrutinib, compared to pLDDT's ρ = 0.03).
摘要: Reliable evaluation of protein structure predictions remains challenging, as metrics like pLDDT capture energetic stability but often miss subtle errors such as atomic clashes or conformational traps reflecting topological frustration within the protein-folding energy landscape. We present CODE (Chain of Diffusion Embeddings), a self-evaluating metric empirically found to quantify topological frustration directly from the latent diffusion embeddings of the AlphaFold3 series of structure predictors in a fully unsupervised manner. Integrating this with pLDDT, we propose CONFIDE, a unified evaluation framework that combines energetic and topological perspectives to improve the reliability of AlphaFold3 and related models. CODE strongly correlates with protein folding rates driven by topological frustration, achieving a correlation of 0.82 compared to pLDDT’s 0.33 (a relative improvement of 148%). CONFIDE significantly enhances the reliability of quality evaluation in molecular glue structure prediction benchmarks, achieving a Spearman correlation of 0.73 with RMSD, compared to pLDDT’s correlation of 0.42, a relative improvement of 73.8%. Beyond quality assessment, our approach applies to diverse drug-design tasks, including all-atom binder design, enzymatic active-site mapping, mutation-induced binding-affinity prediction, nucleic acid aptamer screening, and flexible protein modeling. By combining data-driven embeddings with theoretical insight, CODE and CONFIDE outperform existing metrics across a wide range of biomolecular systems, offering robust and versatile tools to refine structure predictions, advance structural biology, and accelerate drug discovery.