
Paper List
-
Autonomous Agents Coordinating Distributed Discovery Through Emergent Artifact Exchange
This paper addresses the fundamental limitation of current AI-assisted scientific research by enabling truly autonomous, decentralized investigation w...
-
D-MEM: Dopamine-Gated Agentic Memory via Reward Prediction Error Routing
This paper addresses the fundamental scalability bottleneck in LLM agentic memory systems: the O(N²) computational complexity and unbounded API token ...
-
Countershading coloration in blue shark skin emerges from hierarchically organized and spatially tuned photonic architectures inside skin denticles
This paper solves the core problem of how blue sharks achieve their striking dorsoventral countershading camouflage, revealing that coloration origina...
-
Human-like Object Grouping in Self-supervised Vision Transformers
This paper addresses the core challenge of quantifying how well self-supervised vision models capture human-like object grouping in natural scenes, br...
-
Hierarchical pp-Adic Framework for Gene Regulatory Networks: Theory and Stability Analysis
This paper addresses the core challenge of mathematically capturing the inherent hierarchical organization and multi-scale stability of gene regulator...
-
Towards unified brain-to-text decoding across speech production and perception
This paper addresses the core challenge of developing a unified brain-to-text decoding framework that works across both speech production and percepti...
-
Dual-Laws Model for a theory of artificial consciousness
This paper addresses the core challenge of developing a comprehensive, testable theory of consciousness that bridges biological and artificial systems...
-
Pulse desynchronization of neural populations by targeting the centroid of the limit cycle in phase space
This work addresses the core challenge of determining optimal pulse timing and intensity for desynchronizing pathological neural oscillations when the...
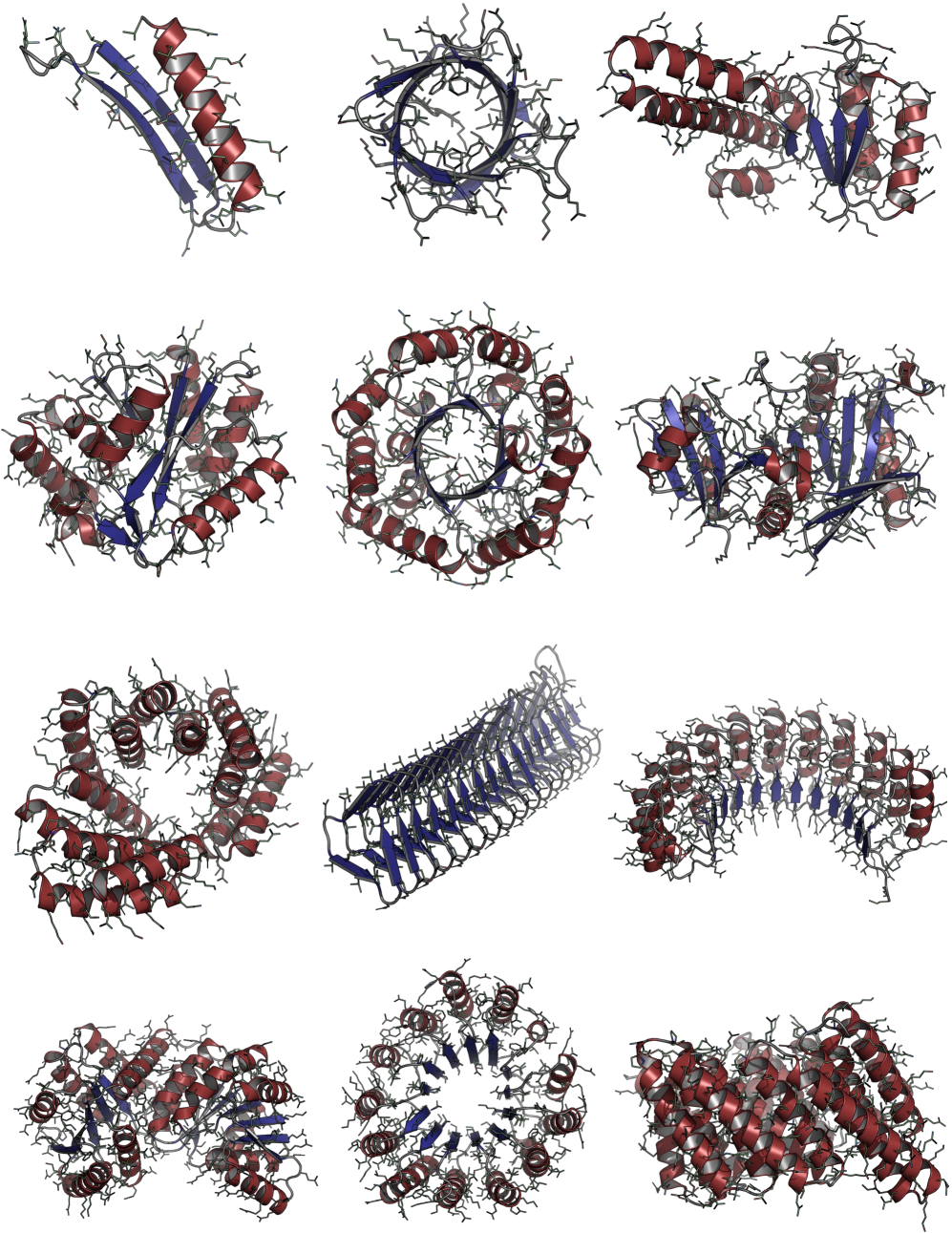
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
NVIDIA | Mila - Quebec AI Institute | Université de Montréal | HEC Montréal | CIFAR AI Chair
30秒速读
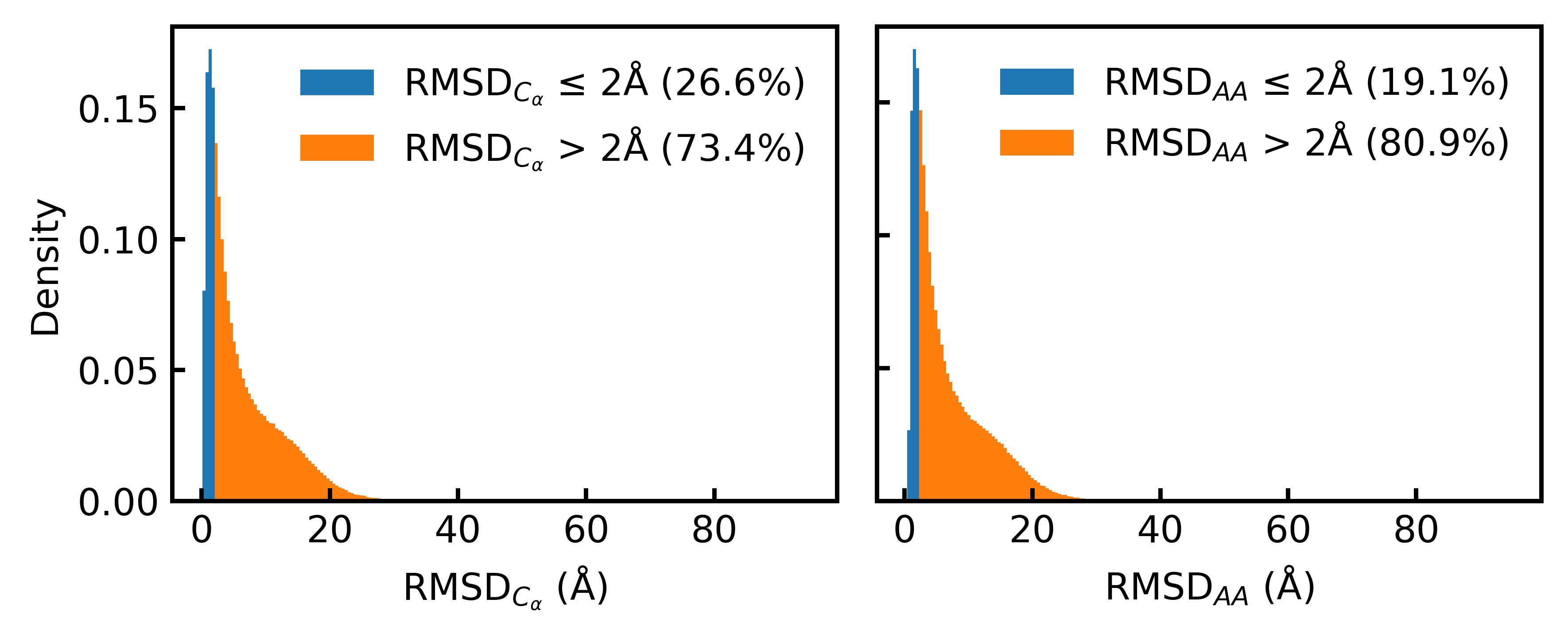
IN SHORT: This paper addresses the core pain point of low sequence-structure alignment in existing synthetic datasets (e.g., AFDB), which severely limits the performance of fully atomistic protein generative models.
核心创新
- Methodology Introduces a novel high-quality synthetic dataset (D_SYN-ours, ~0.46M samples) by leveraging ProteinMPNN for sequence generation and ESMFold for refolding, ensuring aligned and recoverable sequence-structure pairs.
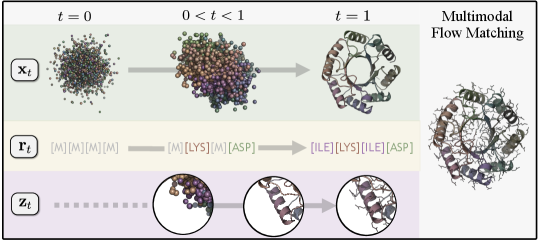
- Methodology Proposes Proteína-Atomística, a unified multi-modal flow-based framework that jointly models the distribution of Cα backbone atoms, discrete amino acid sequences, and non-Cα side-chain atoms in explicit observable space without latent variables.
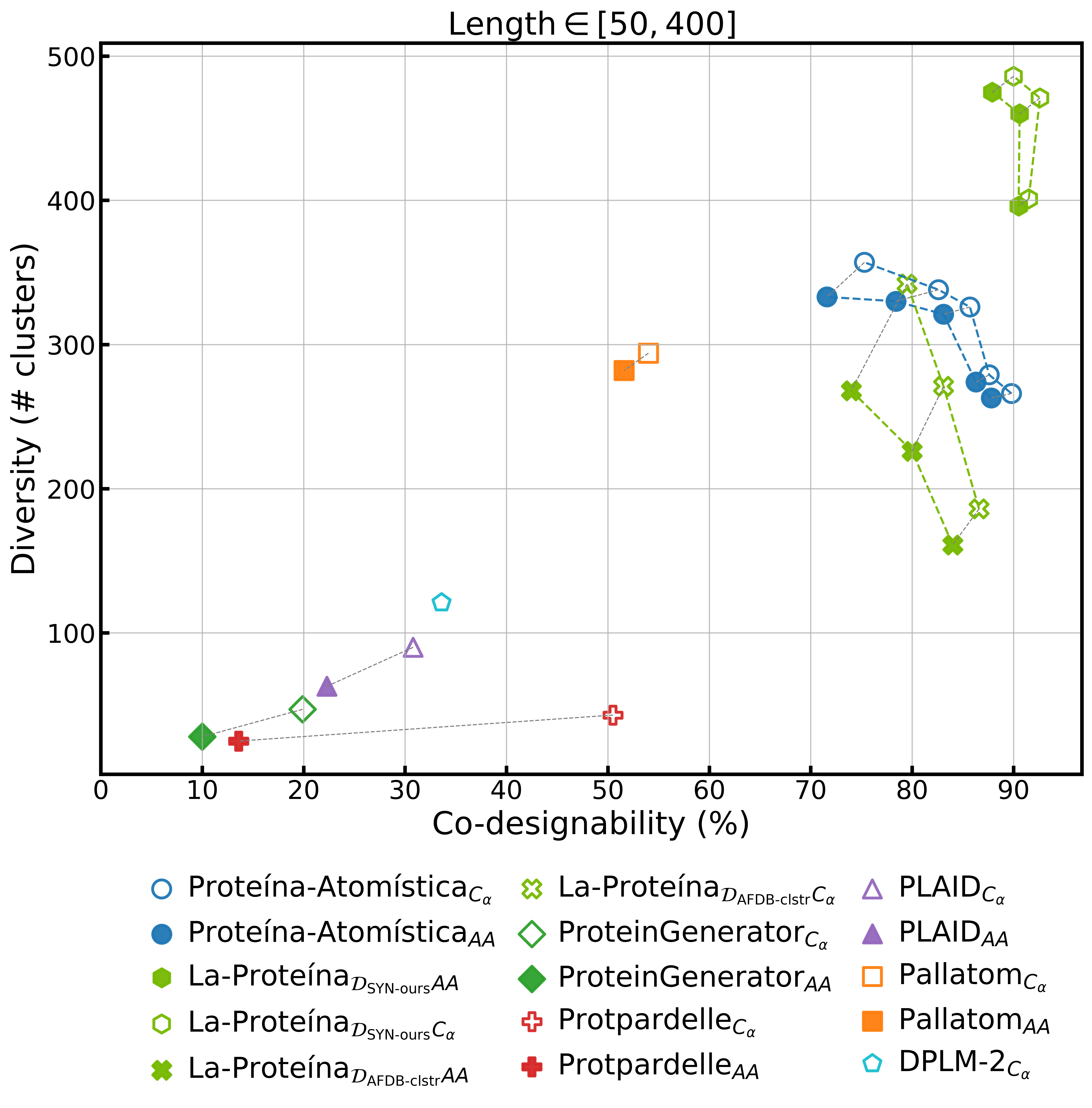
- Biology Demonstrates that consistent synthetic sequences are critical for unlocking structural diversity, with retrained La-Proteína achieving +54% structural diversity and +27% co-designability, and Proteína-Atomística achieving +73% structural diversity and +5% co-designability.
主要结论
- Only 19.1% of the Foldseek-clustered AFDB dataset (D_AFDB-clstr) meets the standard 2Å all-atom RMSD co-designability threshold when refolded with ESMFold, revealing severe sequence-structure misalignment.
- Training on the new aligned dataset D_SYN-ours boosts La-Proteína's performance by +54% in structural diversity and +27% in co-designability, setting a new state-of-the-art.
- The proposed Proteína-Atomística framework, when trained on D_SYN-ours, shows a dramatic +73% improvement in structural diversity and a +5% improvement in co-designability, validating the dataset's broad utility.
摘要: High-quality training datasets are crucial for the development of effective protein design models, but existing synthetic datasets often include unfavorable sequence-structure pairs, impairing generative model performance. We leverage ProteinMPNN, whose sequences are experimentally favorable as well as amenable to folding, together with structure prediction models to align high-quality synthetic structures with recoverable synthetic sequences. In that way, we create a new dataset designed specifically for training expressive, fully atomistic protein generators. By retraining La-Proteína, which models discrete residue type and side chain structure in a continuous latent space, on this dataset, we achieve new state-of-the-art results, with improvements of +54% in structural diversity and +27% in co-designability. To validate the broad utility of our approach, we further introduce Proteína-Atomística, a unified flow-based framework that jointly learns the distribution of protein backbone structure, discrete sequences, and atomistic side chains without latent variables. We again find that training on our new sequence-structure data dramatically boosts benchmark performance, improving Proteína-Atomística’s structural diversity by +73% and co-designability by +5%. Our work highlights the critical importance of aligned sequence-structure data for training high-performance de novo protein design models. All data will be publicly released.