
Paper List
-
SpikGPT: A High-Accuracy and Interpretable Spiking Attention Framework for Single-Cell Annotation
This paper addresses the core challenge of robust single-cell annotation across heterogeneous datasets with batch effects and the critical need to ide...
-
Unlocking hidden biomolecular conformational landscapes in diffusion models at inference time
This paper addresses the core challenge of efficiently and accurately sampling the conformational landscape of biomolecules from diffusion-based struc...
-
Personalized optimization of pediatric HD-tDCS for dose consistency and target engagement
This paper addresses the critical limitation of one-size-fits-all HD-tDCS protocols in pediatric populations by developing a personalized optimization...
-
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein con...
-
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
This paper addresses the core pain point of low sequence-structure alignment in existing synthetic datasets (e.g., AFDB), which severely limits the pe...
-
MoRSAIK: Sequence Motif Reactor Simulation, Analysis and Inference Kit in Python
This work addresses the computational bottleneck in simulating prebiotic RNA reactor dynamics by developing a Python package that tracks sequence moti...
-
On the Approximation of Phylogenetic Distance Functions by Artificial Neural Networks
This paper addresses the core challenge of developing computationally efficient and scalable neural network architectures that can learn accurate phyl...
-
EcoCast: A Spatio-Temporal Model for Continual Biodiversity and Climate Risk Forecasting
This paper addresses the critical bottleneck in conservation: the lack of timely, high-resolution, near-term forecasts of species distribution shifts ...
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
NVIDIA | Mila - Quebec AI Institute | Université de Montréal | HEC Montréal | CIFAR AI Chair
30秒速读
IN SHORT: This paper addresses the core pain point of low sequence-structure alignment in existing synthetic datasets (e.g., AFDB), which severely limits the performance of fully atomistic protein generative models.
核心创新
- Methodology Introduces a novel high-quality synthetic dataset (D_SYN-ours, ~0.46M samples) by leveraging ProteinMPNN for sequence generation and ESMFold for refolding, ensuring aligned and recoverable sequence-structure pairs.
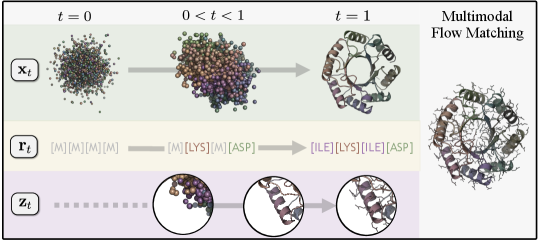
- Methodology Proposes Proteína-Atomística, a unified multi-modal flow-based framework that jointly models the distribution of Cα backbone atoms, discrete amino acid sequences, and non-Cα side-chain atoms in explicit observable space without latent variables.
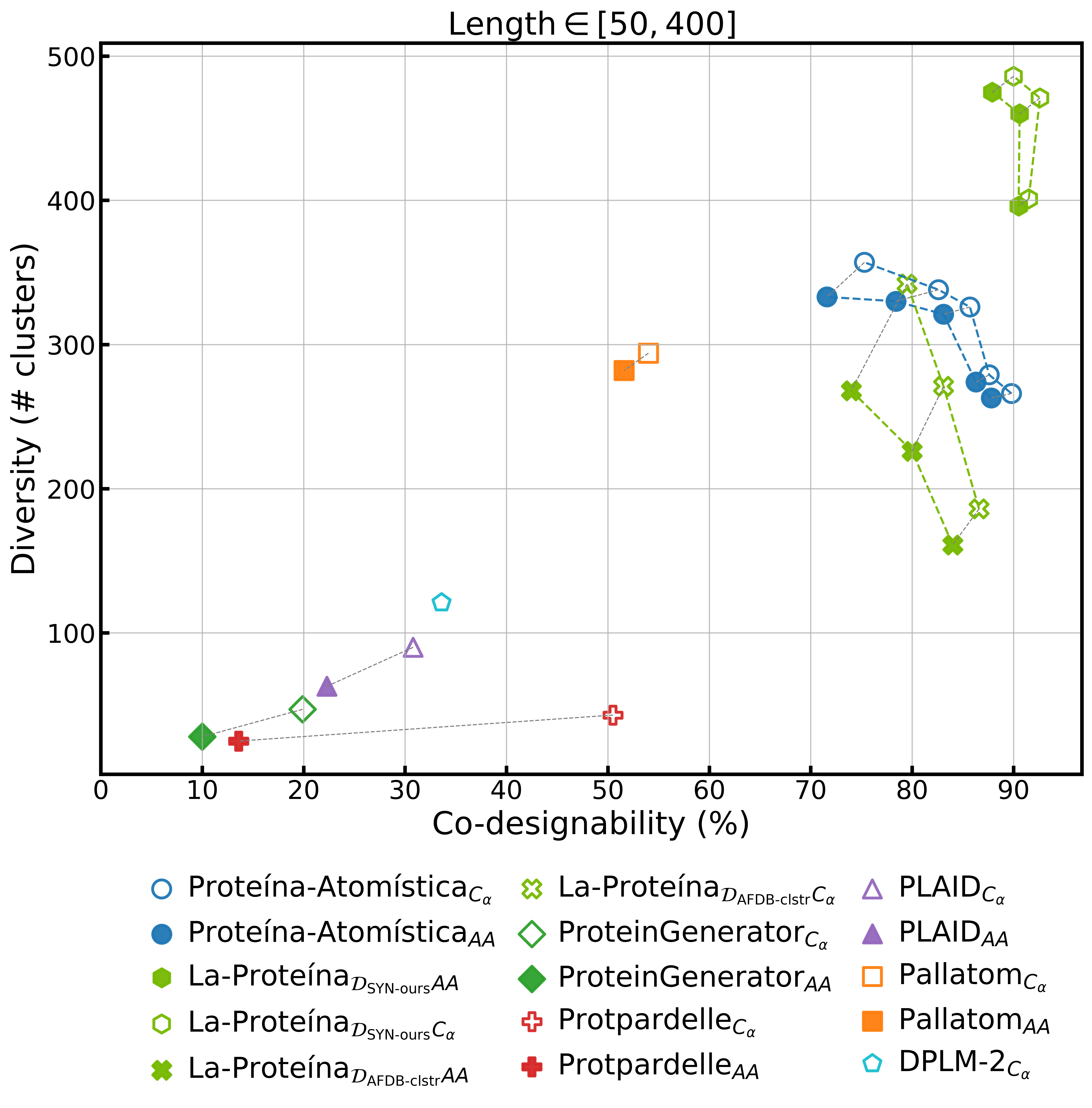
- Biology Demonstrates that consistent synthetic sequences are critical for unlocking structural diversity, with retrained La-Proteína achieving +54% structural diversity and +27% co-designability, and Proteína-Atomística achieving +73% structural diversity and +5% co-designability.
主要结论
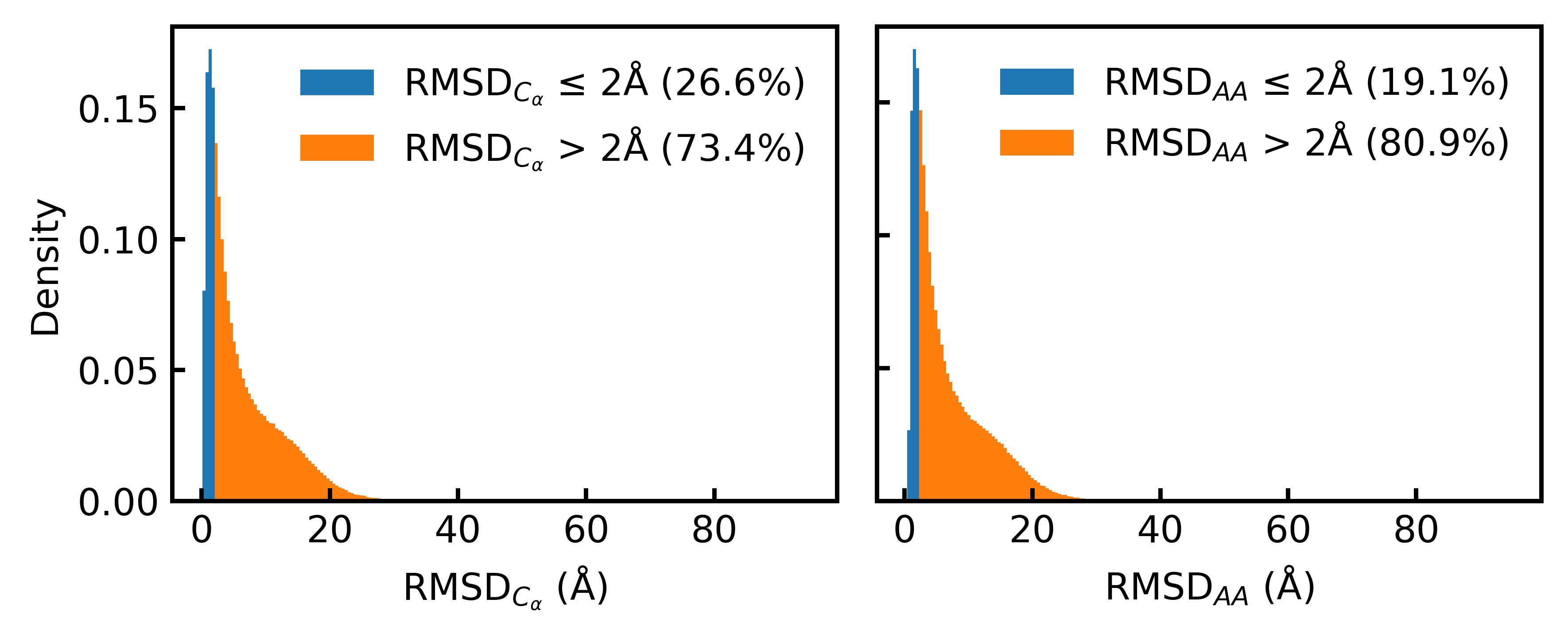
- Only 19.1% of the Foldseek-clustered AFDB dataset (D_AFDB-clstr) meets the standard 2Å all-atom RMSD co-designability threshold when refolded with ESMFold, revealing severe sequence-structure misalignment.
- Training on the new aligned dataset D_SYN-ours boosts La-Proteína's performance by +54% in structural diversity and +27% in co-designability, setting a new state-of-the-art.
- The proposed Proteína-Atomística framework, when trained on D_SYN-ours, shows a dramatic +73% improvement in structural diversity and a +5% improvement in co-designability, validating the dataset's broad utility.
摘要: High-quality training datasets are crucial for the development of effective protein design models, but existing synthetic datasets often include unfavorable sequence-structure pairs, impairing generative model performance. We leverage ProteinMPNN, whose sequences are experimentally favorable as well as amenable to folding, together with structure prediction models to align high-quality synthetic structures with recoverable synthetic sequences. In that way, we create a new dataset designed specifically for training expressive, fully atomistic protein generators. By retraining La-Proteína, which models discrete residue type and side chain structure in a continuous latent space, on this dataset, we achieve new state-of-the-art results, with improvements of +54% in structural diversity and +27% in co-designability. To validate the broad utility of our approach, we further introduce Proteína-Atomística, a unified flow-based framework that jointly learns the distribution of protein backbone structure, discrete sequences, and atomistic side chains without latent variables. We again find that training on our new sequence-structure data dramatically boosts benchmark performance, improving Proteína-Atomística’s structural diversity by +73% and co-designability by +5%. Our work highlights the critical importance of aligned sequence-structure data for training high-performance de novo protein design models. All data will be publicly released.