
Paper List
-
SpikGPT: A High-Accuracy and Interpretable Spiking Attention Framework for Single-Cell Annotation
This paper addresses the core challenge of robust single-cell annotation across heterogeneous datasets with batch effects and the critical need to ide...
-
Unlocking hidden biomolecular conformational landscapes in diffusion models at inference time
This paper addresses the core challenge of efficiently and accurately sampling the conformational landscape of biomolecules from diffusion-based struc...
-
Personalized optimization of pediatric HD-tDCS for dose consistency and target engagement
This paper addresses the critical limitation of one-size-fits-all HD-tDCS protocols in pediatric populations by developing a personalized optimization...
-
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein con...
-
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
This paper addresses the core pain point of low sequence-structure alignment in existing synthetic datasets (e.g., AFDB), which severely limits the pe...
-
MoRSAIK: Sequence Motif Reactor Simulation, Analysis and Inference Kit in Python
This work addresses the computational bottleneck in simulating prebiotic RNA reactor dynamics by developing a Python package that tracks sequence moti...
-
On the Approximation of Phylogenetic Distance Functions by Artificial Neural Networks
This paper addresses the core challenge of developing computationally efficient and scalable neural network architectures that can learn accurate phyl...
-
EcoCast: A Spatio-Temporal Model for Continual Biodiversity and Climate Risk Forecasting
This paper addresses the critical bottleneck in conservation: the lack of timely, high-resolution, near-term forecasts of species distribution shifts ...
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
Department of Computer Science and Genome Center, University of California, Davis | Architecture et Dynamique des Macromolécules Biologiques, UMR 3528 du CNRS, Institut Pasteur | Department of Physics, School of Sciences, Great Bay University | Université Paris-Saclay, CNRS, CEA, Institut de Physique Théorique
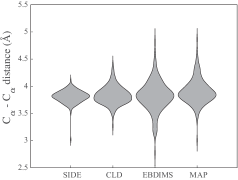
30秒速读
IN SHORT: This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein conformations, a problem where existing methods often produce non-physical trajectories due to oversimplified energy surfaces and steric clashes.
核心创新
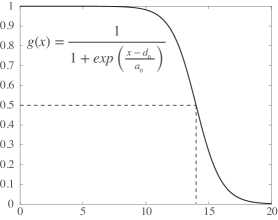
- Methodology Introduces SIDE (Stochastic Integro-Differential Equation), a novel Langevin bridge-based framework that efficiently approximates exact bridge equations at low temperatures to generate constrained transition trajectories.
- Methodology Develops a new coarse-grained potential that combines a Gō-like term (to preserve native backbone geometry) with a Rouse-type elastic energy term (from polymer physics), avoiding the problematic mixing of start/target conformation information used in prior methods like MinActionPath.
- Theory Provides a rigorous stochastic integro-differential formulation derived from the Langevin bridge formalism, which explicitly constrains trajectories to reach a target state within finite time, moving beyond Minimum Action Path (MAP) principles.
主要结论
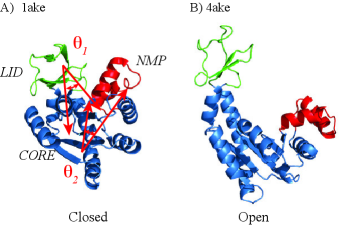
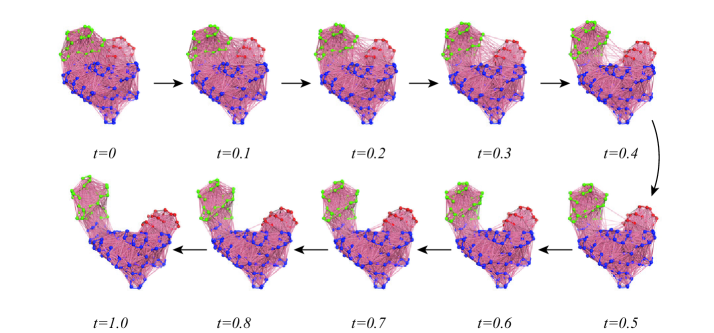
- The SIDE framework generates smooth, low-energy transition trajectories that maintain realistic molecular geometry, as demonstrated on several proteins undergoing large-scale conformational changes.
- SIDE frequently recovers experimentally supported intermediate states along transition paths, suggesting its paths have biological relevance beyond mere endpoint interpolation.
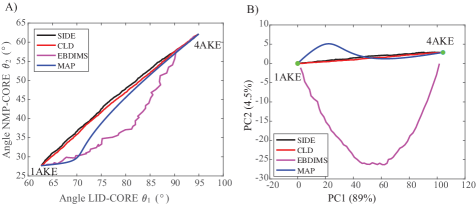
- Compared to established methods like MinActionPath and EBDIMS, SIDE offers improved physical realism and computational efficiency for modeling biomolecular conformational transitions, though challenges remain for highly complex motions.
摘要: We introduce a computational framework for generating realistic transition paths between distinct conformations of large biomolecular systems. The method is built on a stochastic integro-differential formulation derived from the Langevin bridge formalism, which constrains molecular trajectories to reach a prescribed final state within a finite time and yields an efficient low-temperature approximation of the exact bridge equation. To obtain physically meaningful protein transitions, we couple this formulation to a new coarse-grained potential combining a Gō-like term that preserves native backbone geometry with a Rouse-type elastic energy term from polymer physics; we refer to the resulting approach as SIDE. We evaluate SIDE on several proteins undergoing large-scale conformational changes and compare its performance with established methods such as MinActionPath and EBDIMS. SIDE generates smooth, low-energy trajectories that maintain molecular geometry and frequently recover experimentally supported intermediate states. Although challenges remain for highly complex motions—largely due to the simplified coarse-grained potential—our results demonstrate that SIDE offers a powerful and computationally efficient strategy for modeling biomolecular conformational transitions.