
Paper List
-
Developing the PsyCogMetrics™ AI Lab to Evaluate Large Language Models and Advance Cognitive Science
This paper addresses the critical gap between sophisticated LLM evaluation needs and the lack of accessible, scientifically rigorous platforms that in...
-
Equivalence of approximation by networks of single- and multi-spike neurons
This paper resolves the fundamental question of whether single-spike spiking neural networks (SNNs) are inherently less expressive than multi-spike SN...
-
The neuroscience of transformers
提出了Transformer架构与皮层柱微环路之间的新颖计算映射,连接了现代AI与神经科学。
-
Framing local structural identifiability and observability in terms of parameter-state symmetries
This paper addresses the core challenge of systematically determining which parameters and states in a mechanistic ODE model can be uniquely inferred ...
-
Leveraging Phytolith Research using Artificial Intelligence
This paper addresses the critical bottleneck in phytolith research by automating the labor-intensive manual microscopy process through a multimodal AI...
-
Neural network-based encoding in free-viewing fMRI with gaze-aware models
This paper addresses the core challenge of building computationally efficient and ecologically valid brain encoding models for naturalistic vision by ...
-
Scalable DNA Ternary Full Adder Enabled by a Competitive Blocking Circuit
This paper addresses the core bottleneck of carry information attenuation and limited computational scale in DNA binary adders by introducing a scalab...
-
ELISA: An Interpretable Hybrid Generative AI Agent for Expression-Grounded Discovery in Single-Cell Genomics
This paper addresses the critical bottleneck of translating high-dimensional single-cell transcriptomic data into interpretable biological hypotheses ...
Incorporating indel channels into average-case analysis of seed-chain-extend
Carnegie Mellon University, Pittsburgh, PA, USA
30秒速读
IN SHORT: This paper addresses the core pain point of bridging the theoretical gap for the widely used seed-chain-extend heuristic by providing the first rigorous average-case analysis that accounts for insertions and deletions (indels), not just substitutions.
核心创新
- Methodology Introduces a generalized definition of 'recoverability' and a 'homologous path' to mathematically model the correct alignment under indel mutation channels, moving beyond the simpler 'homologous diagonal' used for substitutions only.
- Theory Develops new mathematical machinery to handle the dependence structure of neighboring anchors and the existence of 'clipping anchors' (partially correct anchors), which are unique challenges introduced by indels.
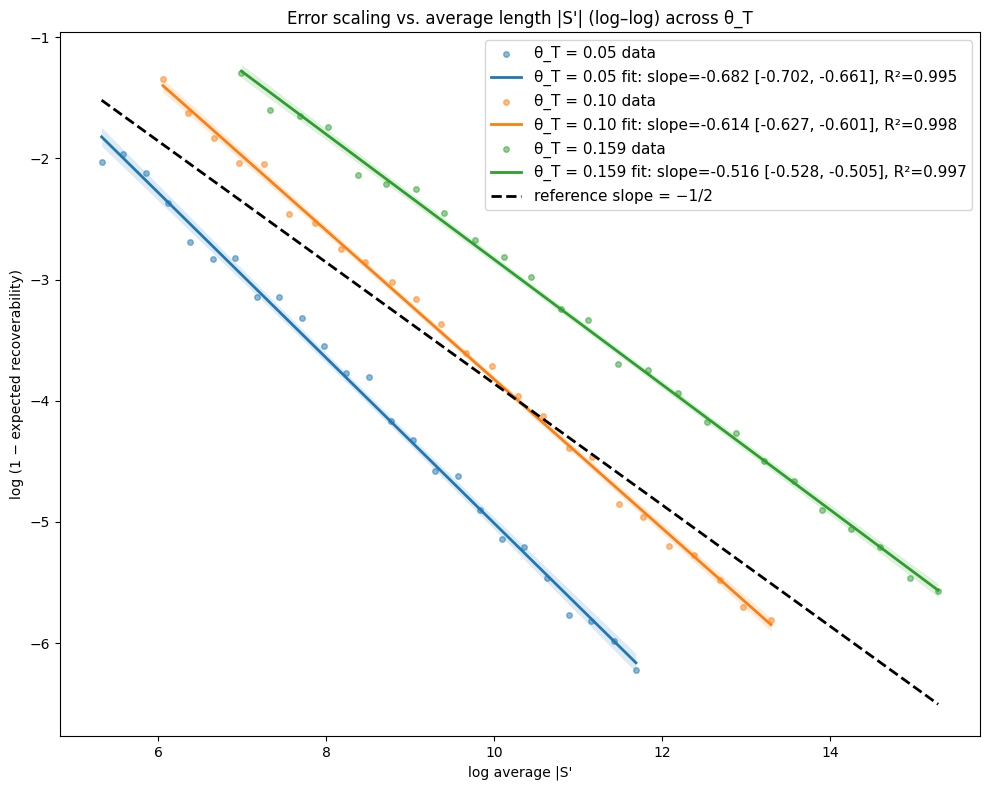
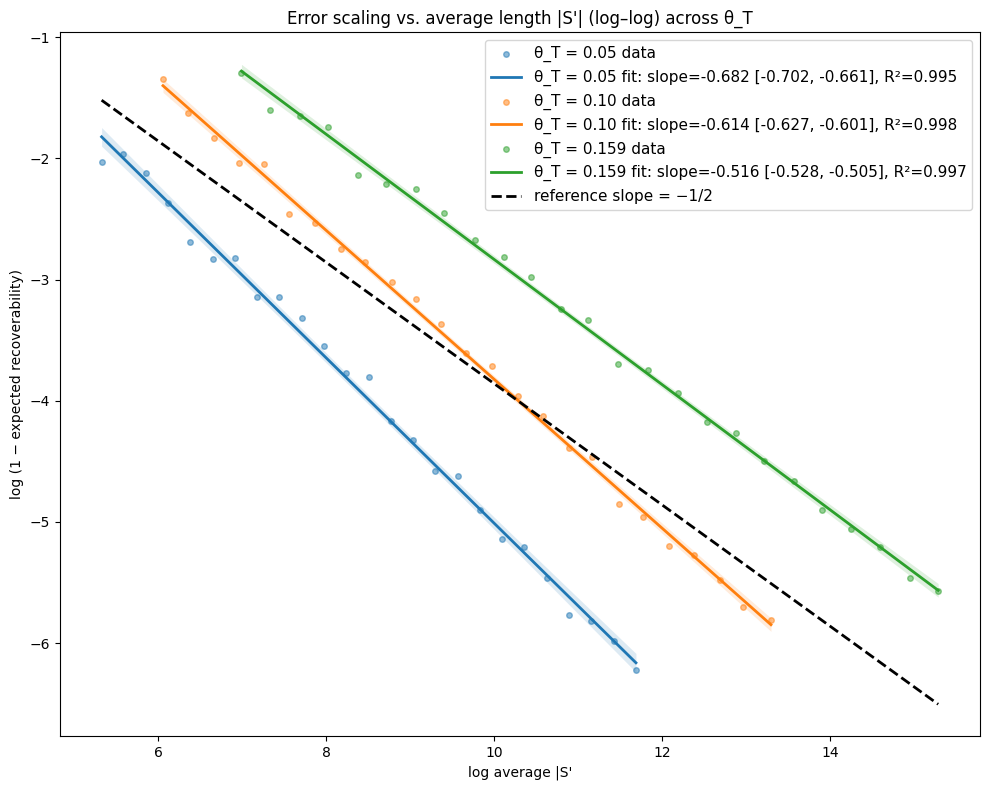
- Theory Proves that under a total mutation rate θ_T < 0.159, optimal linear-gap cost chaining achieves an expected recoverability of ≥ 1 - O(1/√m), generalizing the prior substitution-only result to a biologically realistic model.
主要结论
- The expected recoverability of an optimal chain under linear-gap cost chaining is ≥ 1 - O(1/√m) when the total mutation rate θ_T (sum of substitution, insertion, deletion rates) is less than 0.159.
- The expected runtime of the algorithm is O(m n^(3.15·θ_T) log n). For example, at a θ_T of 0.05 (similar to human-chimp divergence), the exponent is ~1.12, leading to near-linear scaling.
- The analysis successfully bridges theory and practice by extending the proof framework to handle indels, justifying the heuristic's empirical effectiveness on real genomic data which contains indels.
摘要: Given a sequence s1 of n letters drawn i.i.d. from an alphabet of size σ and a mutated substring s2 of length m<n, we often want to recover the mutation history that generated s2 from s1. Modern sequence aligners are widely used for this task, and many employ the seed-chain-extend heuristic with k-mer seeds. Previously, Shaw and Yu showed that optimal linear-gap cost chaining can produce a chain with 1−O(1/m) recoverability, the proportion of the mutation history that is recovered, in O(mn^(2.43θ) log n) expected time, where θ<0.206 is the mutation rate under a substitution-only channel and s1 is assumed to be uniformly random. However, a gap remains between theory and practice, since real genomic data includes insertions and deletions (indels), and yet seed-chain-extend remains effective. In this paper, we generalize those prior results by introducing mathematical machinery to deal with the two new obstacles introduced by indel channels: the dependence of neighboring anchors and the presence of anchors that are only partially correct. We are thus able to prove that the expected recoverability of an optimal chain is ≥1−O(1/√m) and the expected runtime is O(mn^(3.15·θ_T) log n), when the total mutation rate given by the sum of the substitution, insertion, and deletion mutation rates (θ_T = θ_i + θ_d + θ_s) is less than 0.159.