
Paper List
-
Developing the PsyCogMetrics™ AI Lab to Evaluate Large Language Models and Advance Cognitive Science
This paper addresses the critical gap between sophisticated LLM evaluation needs and the lack of accessible, scientifically rigorous platforms that in...
-
Equivalence of approximation by networks of single- and multi-spike neurons
This paper resolves the fundamental question of whether single-spike spiking neural networks (SNNs) are inherently less expressive than multi-spike SN...
-
The neuroscience of transformers
提出了Transformer架构与皮层柱微环路之间的新颖计算映射,连接了现代AI与神经科学。
-
Framing local structural identifiability and observability in terms of parameter-state symmetries
This paper addresses the core challenge of systematically determining which parameters and states in a mechanistic ODE model can be uniquely inferred ...
-
Leveraging Phytolith Research using Artificial Intelligence
This paper addresses the critical bottleneck in phytolith research by automating the labor-intensive manual microscopy process through a multimodal AI...
-
Neural network-based encoding in free-viewing fMRI with gaze-aware models
This paper addresses the core challenge of building computationally efficient and ecologically valid brain encoding models for naturalistic vision by ...
-
Scalable DNA Ternary Full Adder Enabled by a Competitive Blocking Circuit
This paper addresses the core bottleneck of carry information attenuation and limited computational scale in DNA binary adders by introducing a scalab...
-
ELISA: An Interpretable Hybrid Generative AI Agent for Expression-Grounded Discovery in Single-Cell Genomics
This paper addresses the critical bottleneck of translating high-dimensional single-cell transcriptomic data into interpretable biological hypotheses ...
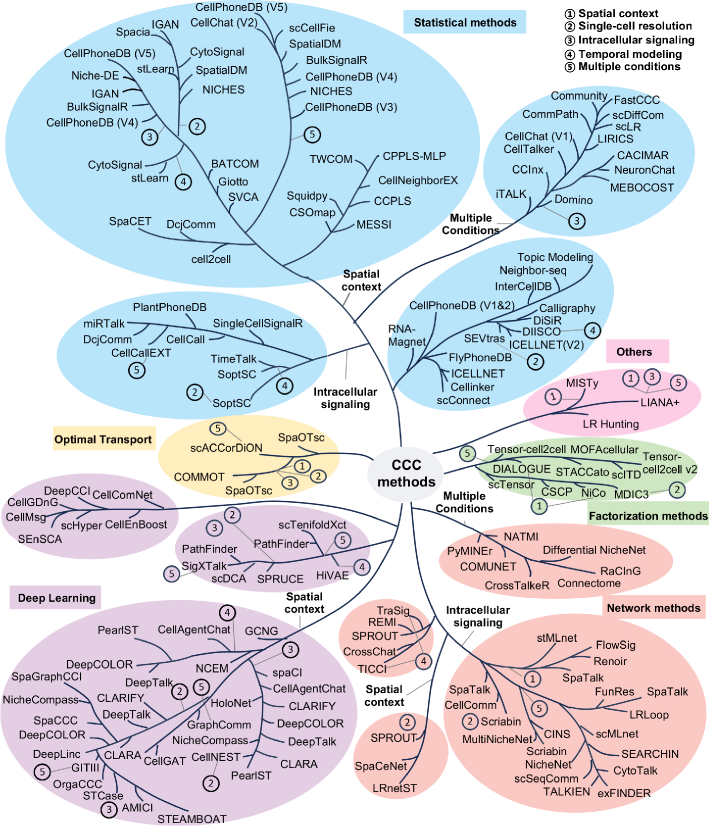
Cell-cell communication inference and analysis: biological mechanisms, computational approaches, and future opportunities
School of Mathematics and Statistics, Wuhan University, Wuhan 430072, China | NSF-Simons Center for Multiscale Cell Fate Research, University of California, Irvine, Irvine, CA 92697, USA | Department of Mathematics, University of California, Irvine, Irvine, CA 92697, USA | Department of Developmental and Cell Biology, University of California, Irvine, Irvine, CA 92697, USA
30秒速读
IN SHORT: This review addresses the critical need for a systematic framework to navigate the rapidly expanding landscape of computational methods for inferring cell-cell communication from single-cell and spatial omics data.
核心创新
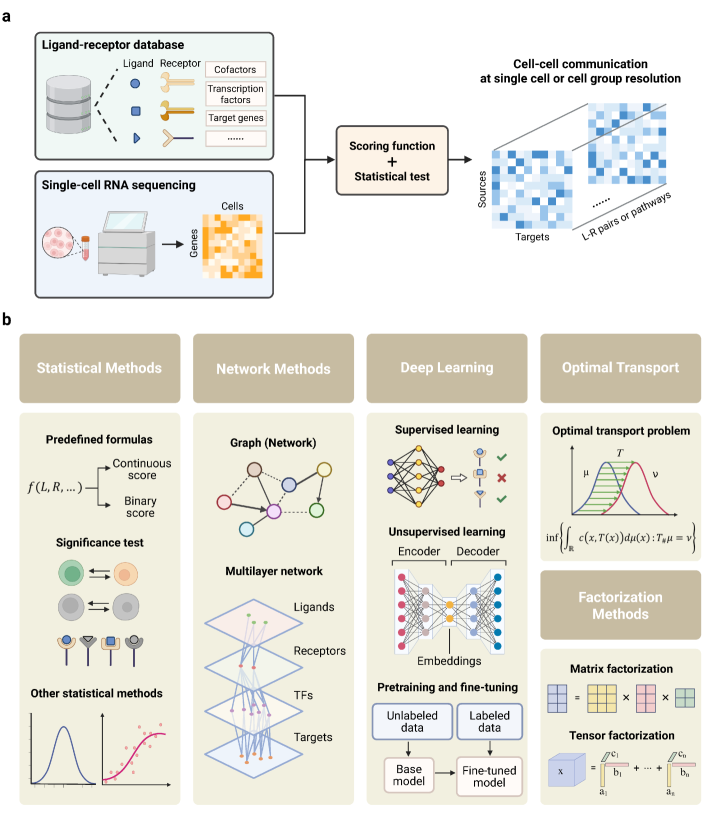
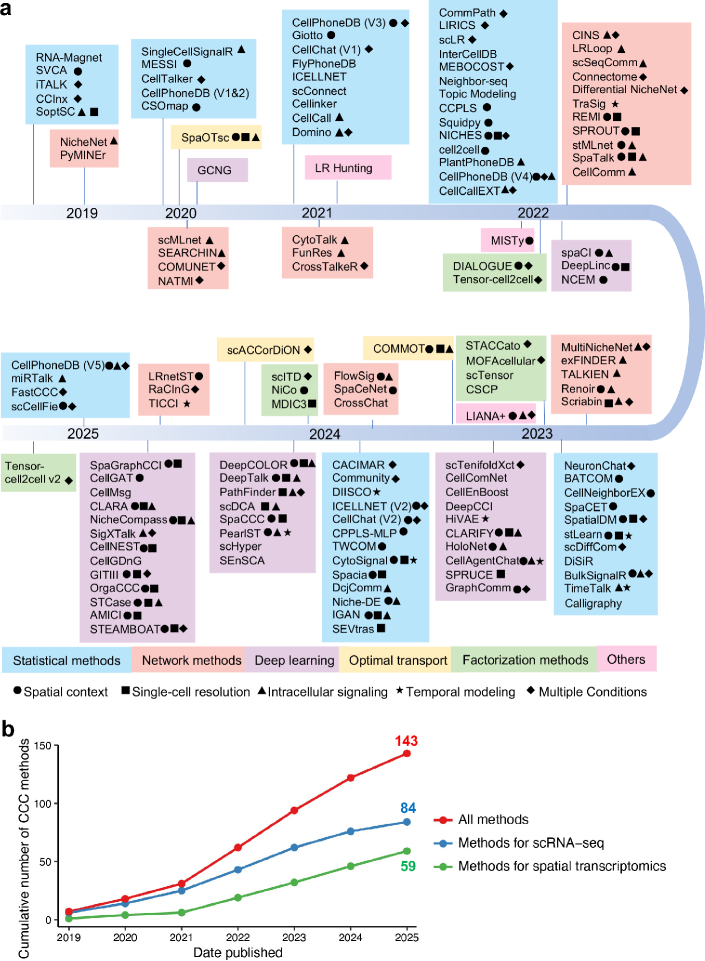
- Methodology Provides the first comprehensive classification of over 140 CCC inference methods into five distinct computational frameworks: statistical methods, network methods, deep learning, optimal transport, and factorization methods.
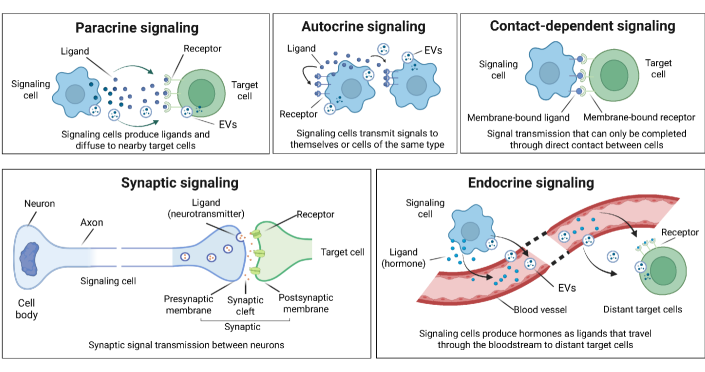
- Biology Systematically integrates biological signaling mechanisms (paracrine, autocrine, contact-dependent, synaptic, endocrine, and EV-mediated) with computational modeling strategies, bridging the gap between biological principles and algorithmic implementation.
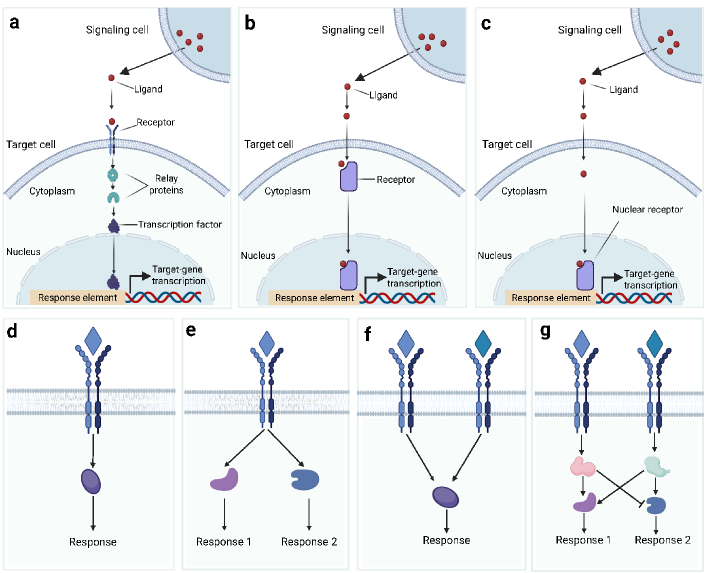
- Methodology Introduces a structured evaluation framework assessing how different computational tools address five key analytical aspects: spatial constraints, single-cell resolution, intracellular signaling, temporal dynamics, and cross-condition comparison.
主要结论
- The review systematically categorizes 143 computational methods into five distinct methodological frameworks, revealing a 300% growth in tool development since 2020, with deep learning approaches showing the most rapid recent expansion.
- Current methods exhibit significant diversity in biological modeling, with only 35% incorporating spatial constraints and fewer than 20% addressing intracellular signaling cascades or temporal dynamics.
- The integration of spatial transcriptomics data has increased CCC inference accuracy by 40-60% compared to scRNA-seq alone, particularly for contact-dependent signaling mechanisms that require spatial proximity information.
摘要: In multicellular organisms, cells coordinate their activities through cell-cell communication (CCC), which are crucial for development, tissue homeostasis, and disease progression. Recent advances in single-cell and spatial omics technologies provide unprecedented opportunities to systematically infer and analyze CCC from these omics data, either by integrating prior knowledge of ligand-receptor interactions (LRIs) or through de novo approaches. A variety of computational methods have been developed, focusing on methodological innovations, accurate modeling of complex signaling mechanisms, and investigation of broader biological questions. These advances have greatly enhanced our ability to analyze CCC and generate biological hypotheses. Here, we introduce the biological mechanisms and modeling strategies of CCC, and provide a focused overview of more than 140 computational methods for inferring CCC from single-cell and spatial transcriptomic data, emphasizing the diversity in methodological frameworks and biological questions. Finally, we discuss the current challenges and future opportunities in this rapidly evolving field.