
Paper List
-
Autonomous Agents Coordinating Distributed Discovery Through Emergent Artifact Exchange
This paper addresses the fundamental limitation of current AI-assisted scientific research by enabling truly autonomous, decentralized investigation w...
-
D-MEM: Dopamine-Gated Agentic Memory via Reward Prediction Error Routing
This paper addresses the fundamental scalability bottleneck in LLM agentic memory systems: the O(N²) computational complexity and unbounded API token ...
-
Countershading coloration in blue shark skin emerges from hierarchically organized and spatially tuned photonic architectures inside skin denticles
This paper solves the core problem of how blue sharks achieve their striking dorsoventral countershading camouflage, revealing that coloration origina...
-
Human-like Object Grouping in Self-supervised Vision Transformers
This paper addresses the core challenge of quantifying how well self-supervised vision models capture human-like object grouping in natural scenes, br...
-
Hierarchical pp-Adic Framework for Gene Regulatory Networks: Theory and Stability Analysis
This paper addresses the core challenge of mathematically capturing the inherent hierarchical organization and multi-scale stability of gene regulator...
-
Towards unified brain-to-text decoding across speech production and perception
This paper addresses the core challenge of developing a unified brain-to-text decoding framework that works across both speech production and percepti...
-
Dual-Laws Model for a theory of artificial consciousness
This paper addresses the core challenge of developing a comprehensive, testable theory of consciousness that bridges biological and artificial systems...
-
Pulse desynchronization of neural populations by targeting the centroid of the limit cycle in phase space
This work addresses the core challenge of determining optimal pulse timing and intensity for desynchronizing pathological neural oscillations when the...
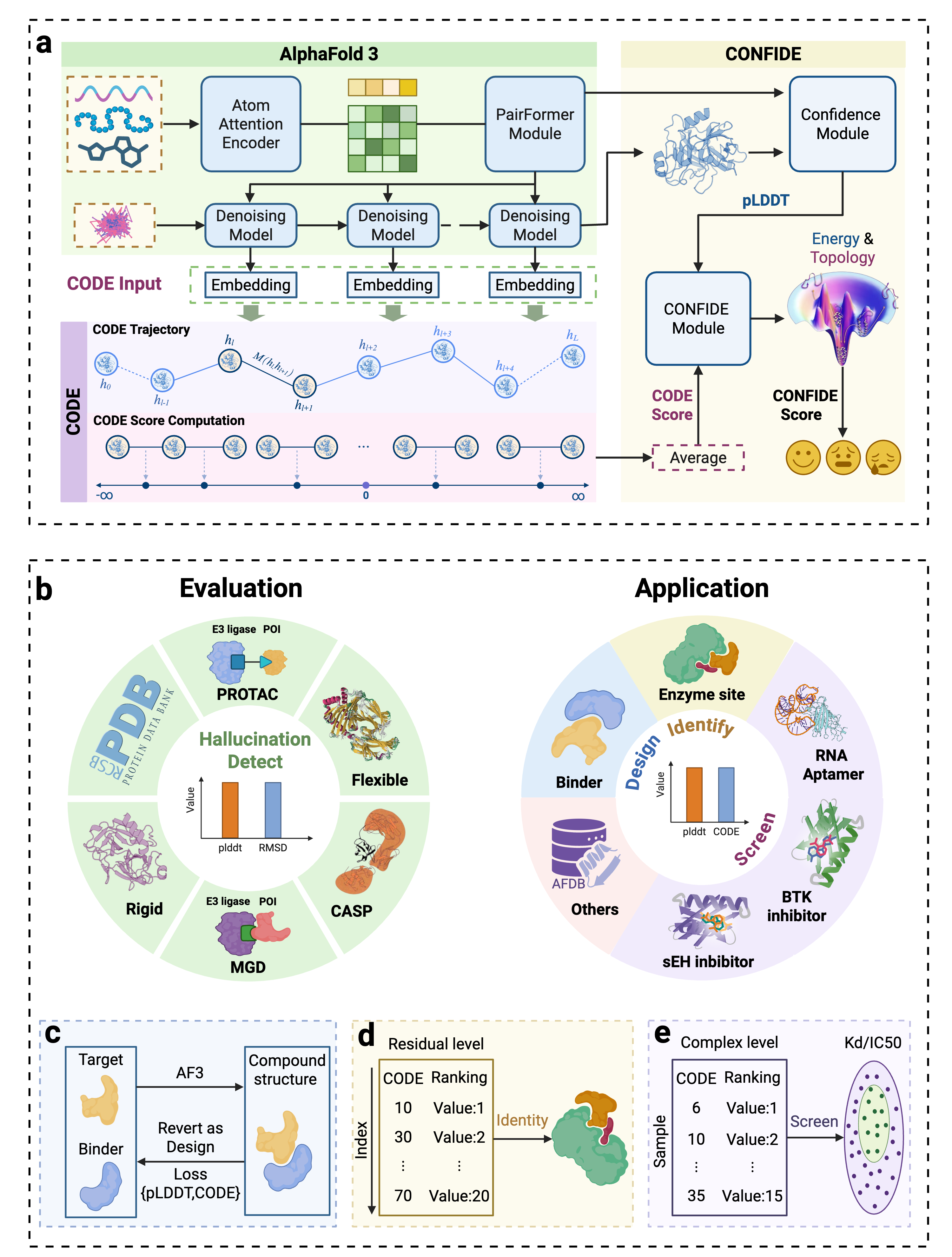
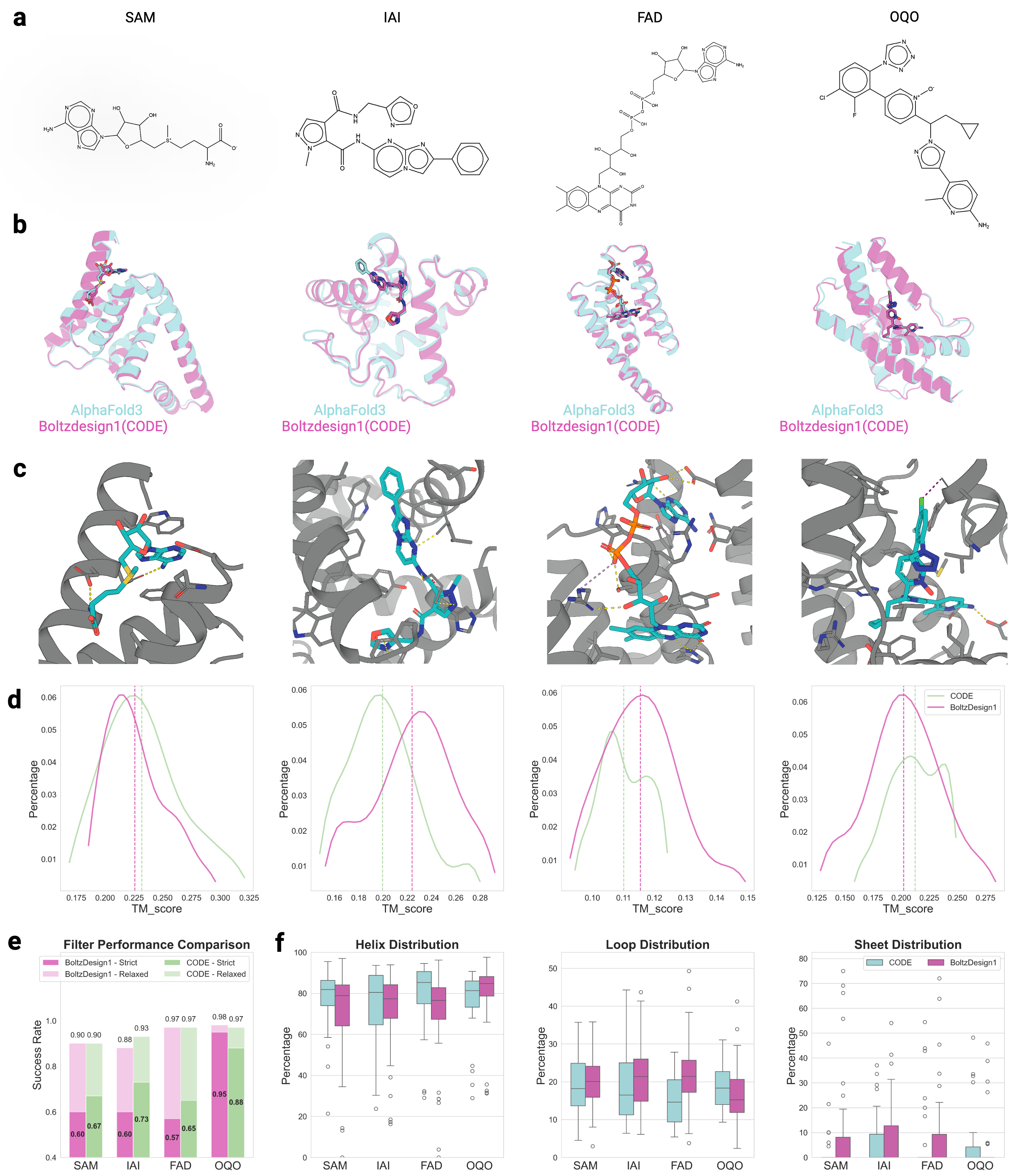
CONFIDE: Hallucination Assessment for Reliable Biomolecular Structure Prediction and Design
The Chinese University of Hong Kong | Zhejiang University | Macao Polytechnic University | University of Electronic Science and Technology of China
30秒速读
IN SHORT: This paper addresses the critical limitation of current protein structure prediction models (like AlphaFold3) where high-confidence scores (pLDDT) can be misleading, failing to detect subtle structural errors like atomic clashes and topological traps, which undermines reliability in downstream applications like drug discovery.
核心创新
- Methodology Introduces CODE (Chain of Diffusion Embeddings), a novel, unsupervised metric derived from AlphaFold3's latent diffusion embeddings that directly quantifies topological frustration, a key factor in protein folding kinetics previously overlooked by confidence scores.
- Methodology Proposes CONFIDE, a unified evaluation framework that integrates the energetic perspective of pLDDT with the topological perspective of CODE, providing a more comprehensive and reliable assessment of predicted biomolecular structures.
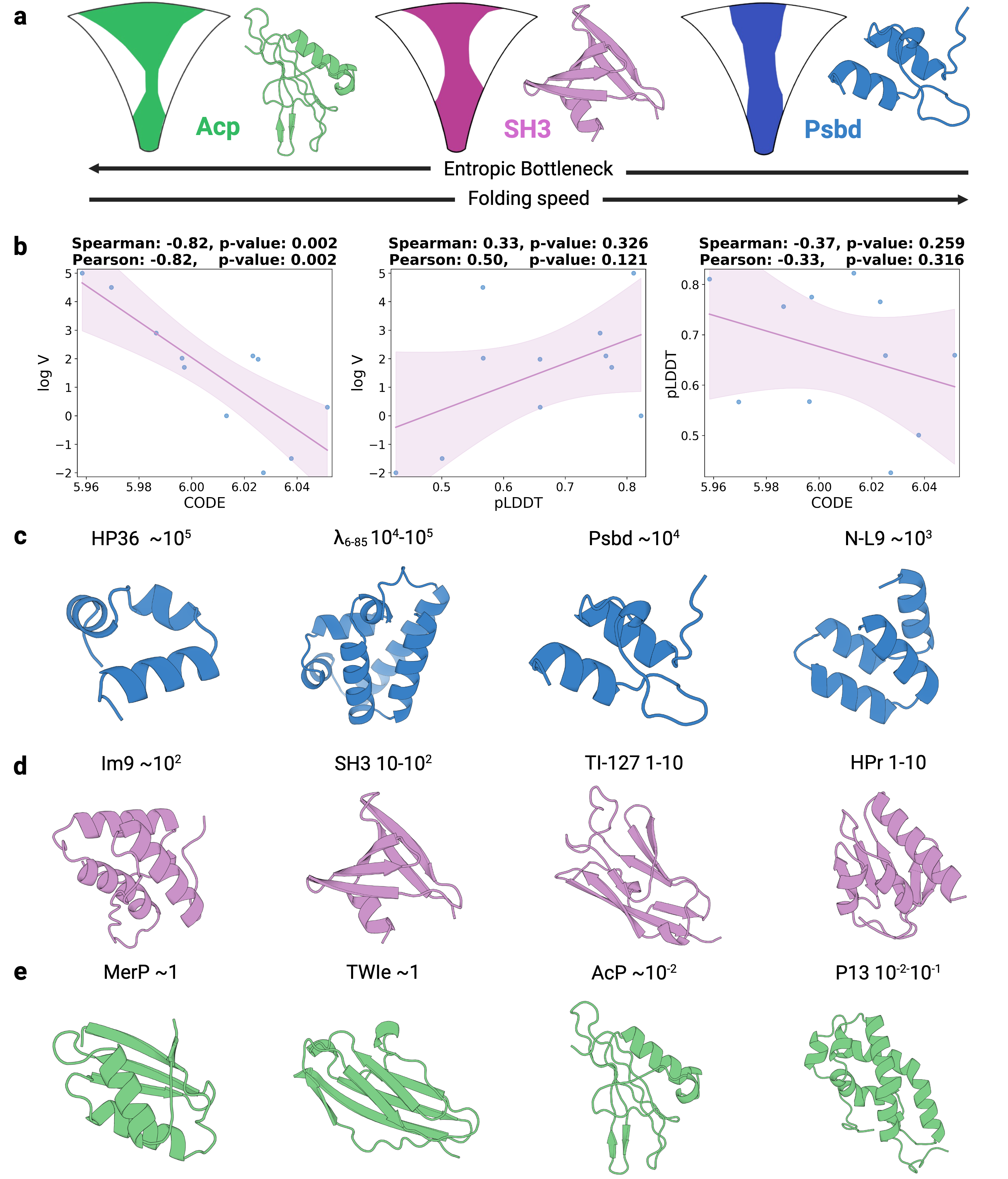
- Biology Establishes a strong empirical link between the CODE metric and protein folding rates driven by topological frustration (Spearman correlation of -0.82, p=0.002), offering a data-driven proxy for a complex biophysical phenomenon.
主要结论
- CODE demonstrates a strong, statistically significant correlation with protein folding rates mediated by topological frustration (Spearman ρ = -0.82, p=0.002), far outperforming pLDDT (ρ = 0.33, p=0.326).
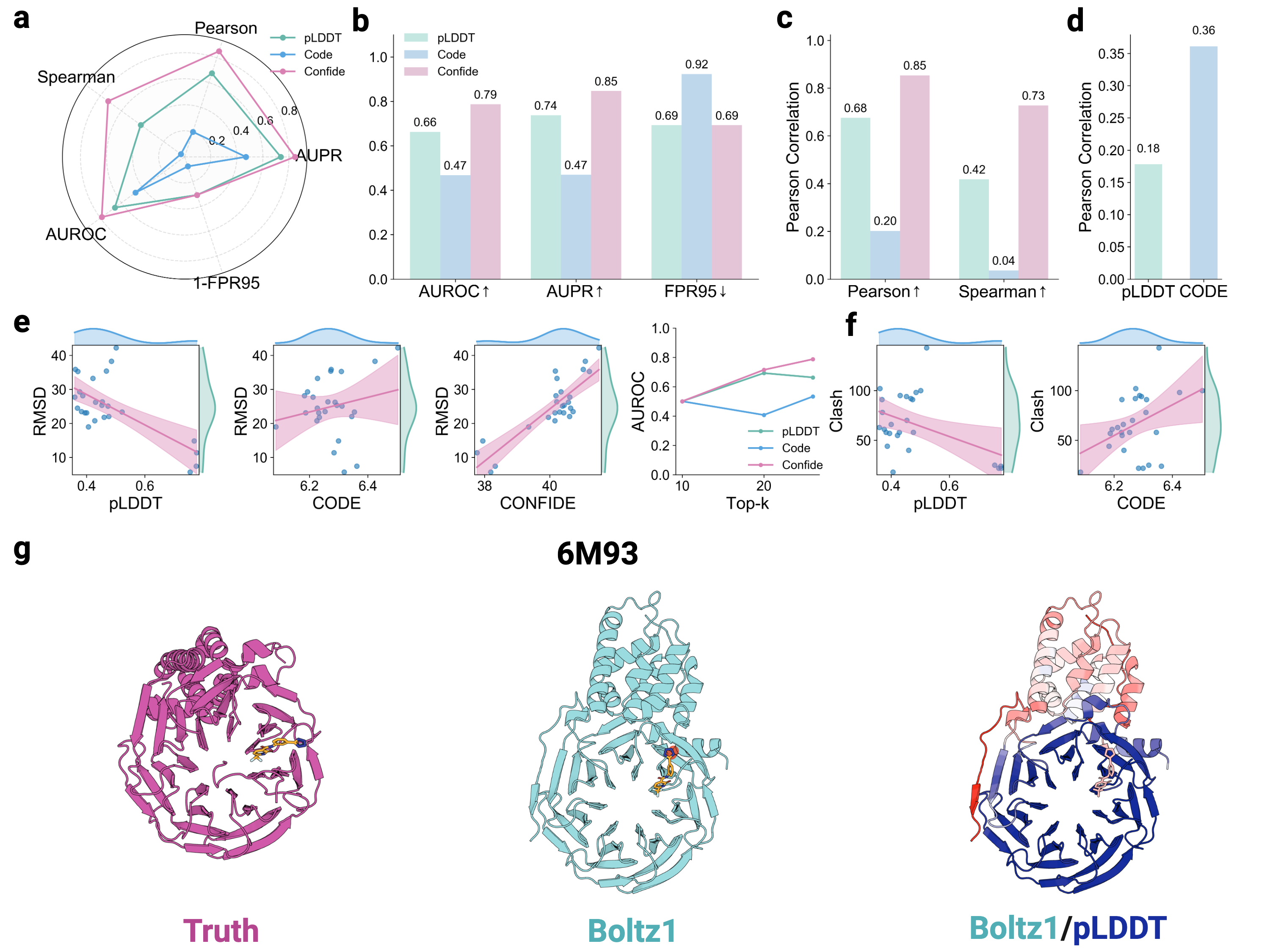
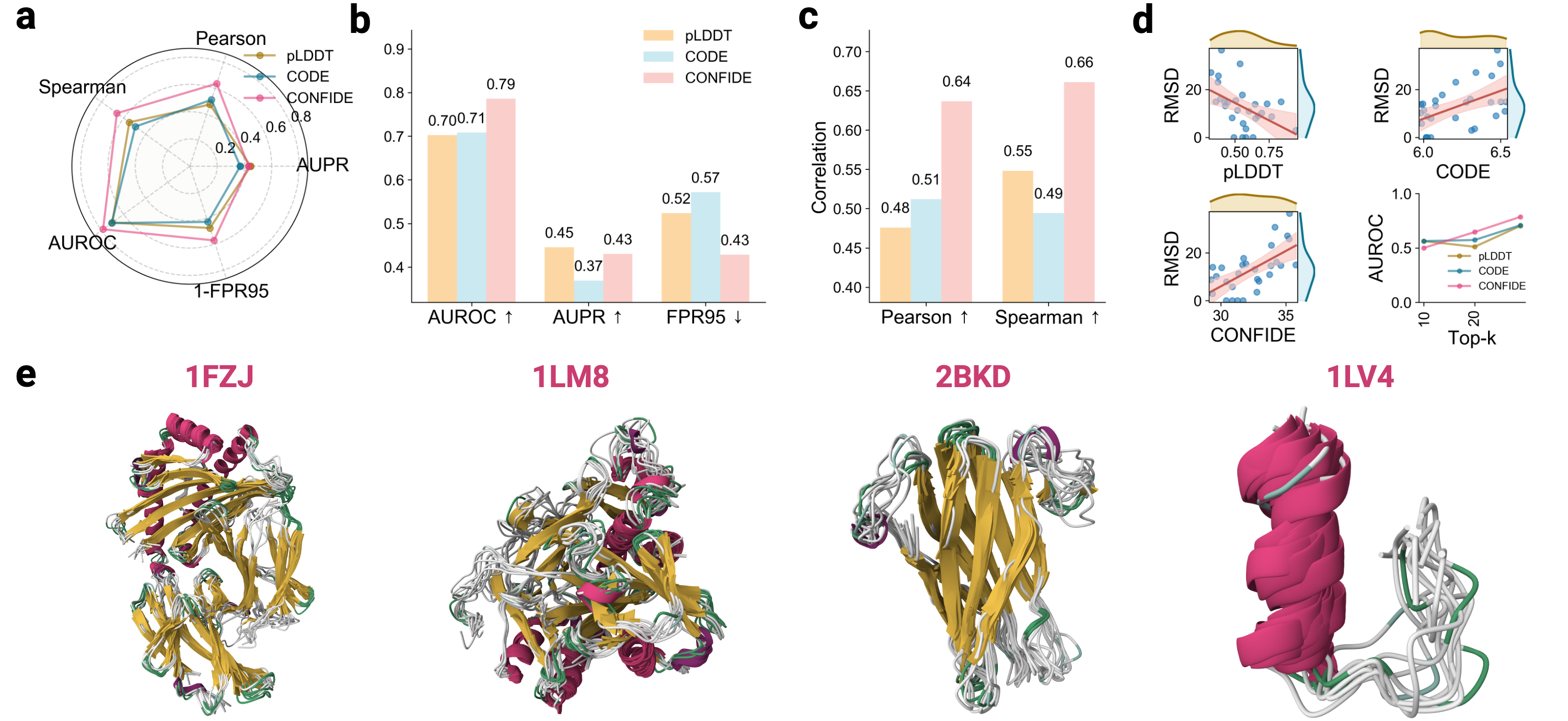
- The CONFIDE framework significantly improves hallucination detection, achieving a Spearman correlation of 0.73 with RMSD on molecular glue benchmarks, a 73.8% relative improvement over pLDDT's correlation of 0.42.
- CONFIDE enables practical downstream applications, improving binder design success rates (e.g., +13% for IAI) and accurately predicting mutation-induced binding affinity changes (Spearman ρ = 0.83 for BTK vs. Fenebrutinib, compared to pLDDT's ρ = 0.03).
摘要: Reliable evaluation of protein structure predictions remains challenging, as metrics like pLDDT capture energetic stability but often miss subtle errors such as atomic clashes or conformational traps reflecting topological frustration within the protein-folding energy landscape. We present CODE (Chain of Diffusion Embeddings), a self-evaluating metric empirically found to quantify topological frustration directly from the latent diffusion embeddings of the AlphaFold3 series of structure predictors in a fully unsupervised manner. Integrating this with pLDDT, we propose CONFIDE, a unified evaluation framework that combines energetic and topological perspectives to improve the reliability of AlphaFold3 and related models. CODE strongly correlates with protein folding rates driven by topological frustration, achieving a correlation of 0.82 compared to pLDDT’s 0.33 (a relative improvement of 148%). CONFIDE significantly enhances the reliability of quality evaluation in molecular glue structure prediction benchmarks, achieving a Spearman correlation of 0.73 with RMSD, compared to pLDDT’s correlation of 0.42, a relative improvement of 73.8%. Beyond quality assessment, our approach applies to diverse drug-design tasks, including all-atom binder design, enzymatic active-site mapping, mutation-induced binding-affinity prediction, nucleic acid aptamer screening, and flexible protein modeling. By combining data-driven embeddings with theoretical insight, CODE and CONFIDE outperform existing metrics across a wide range of biomolecular systems, offering robust and versatile tools to refine structure predictions, advance structural biology, and accelerate drug discovery.