
Paper List
-
Developing the PsyCogMetrics™ AI Lab to Evaluate Large Language Models and Advance Cognitive Science
This paper addresses the critical gap between sophisticated LLM evaluation needs and the lack of accessible, scientifically rigorous platforms that in...
-
Equivalence of approximation by networks of single- and multi-spike neurons
This paper resolves the fundamental question of whether single-spike spiking neural networks (SNNs) are inherently less expressive than multi-spike SN...
-
The neuroscience of transformers
提出了Transformer架构与皮层柱微环路之间的新颖计算映射,连接了现代AI与神经科学。
-
Framing local structural identifiability and observability in terms of parameter-state symmetries
This paper addresses the core challenge of systematically determining which parameters and states in a mechanistic ODE model can be uniquely inferred ...
-
Leveraging Phytolith Research using Artificial Intelligence
This paper addresses the critical bottleneck in phytolith research by automating the labor-intensive manual microscopy process through a multimodal AI...
-
Neural network-based encoding in free-viewing fMRI with gaze-aware models
This paper addresses the core challenge of building computationally efficient and ecologically valid brain encoding models for naturalistic vision by ...
-
Scalable DNA Ternary Full Adder Enabled by a Competitive Blocking Circuit
This paper addresses the core bottleneck of carry information attenuation and limited computational scale in DNA binary adders by introducing a scalab...
-
ELISA: An Interpretable Hybrid Generative AI Agent for Expression-Grounded Discovery in Single-Cell Genomics
This paper addresses the critical bottleneck of translating high-dimensional single-cell transcriptomic data into interpretable biological hypotheses ...
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
NVIDIA | Mila - Quebec AI Institute | Université de Montréal | HEC Montréal | CIFAR AI Chair
30秒速读
IN SHORT: This paper addresses the core pain point of low sequence-structure alignment in existing synthetic datasets (e.g., AFDB), which severely limits the performance of fully atomistic protein generative models.
核心创新
- Methodology Introduces a novel high-quality synthetic dataset (D_SYN-ours, ~0.46M samples) by leveraging ProteinMPNN for sequence generation and ESMFold for refolding, ensuring aligned and recoverable sequence-structure pairs.
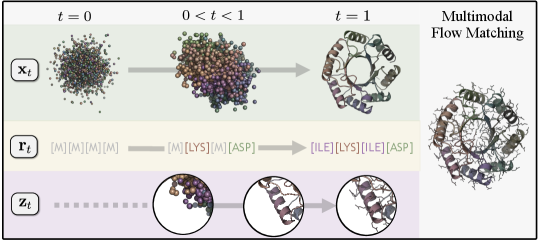
- Methodology Proposes Proteína-Atomística, a unified multi-modal flow-based framework that jointly models the distribution of Cα backbone atoms, discrete amino acid sequences, and non-Cα side-chain atoms in explicit observable space without latent variables.
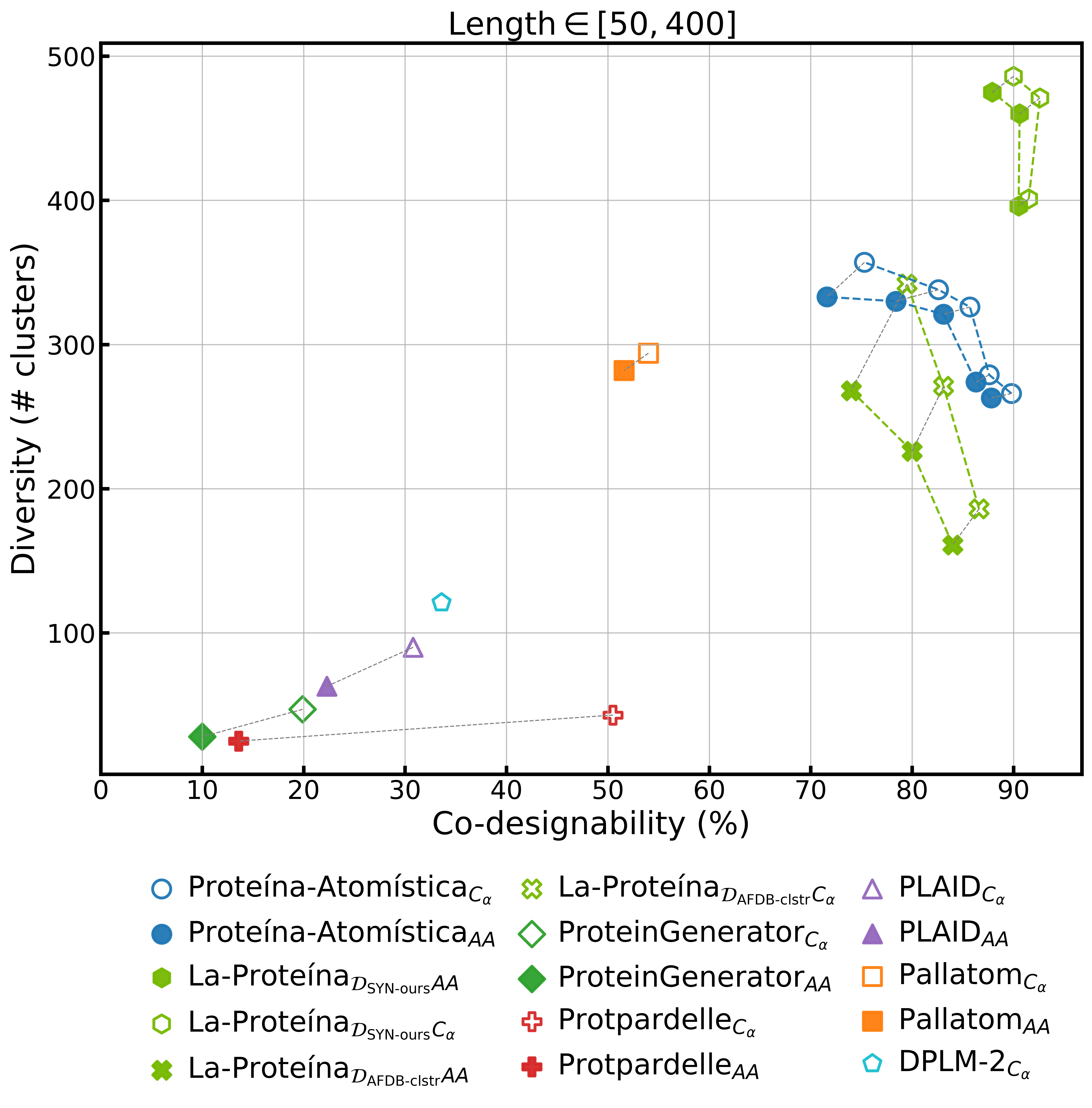
- Biology Demonstrates that consistent synthetic sequences are critical for unlocking structural diversity, with retrained La-Proteína achieving +54% structural diversity and +27% co-designability, and Proteína-Atomística achieving +73% structural diversity and +5% co-designability.
主要结论
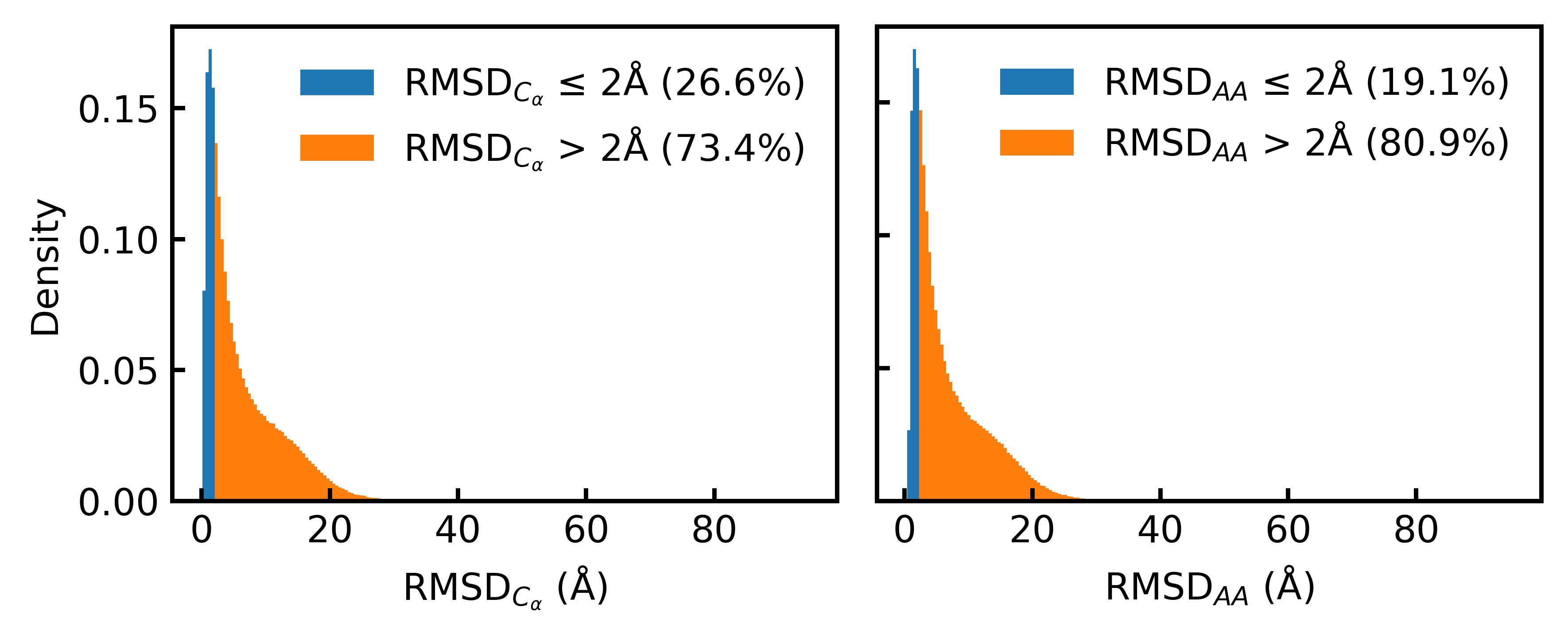
- Only 19.1% of the Foldseek-clustered AFDB dataset (D_AFDB-clstr) meets the standard 2Å all-atom RMSD co-designability threshold when refolded with ESMFold, revealing severe sequence-structure misalignment.
- Training on the new aligned dataset D_SYN-ours boosts La-Proteína's performance by +54% in structural diversity and +27% in co-designability, setting a new state-of-the-art.
- The proposed Proteína-Atomística framework, when trained on D_SYN-ours, shows a dramatic +73% improvement in structural diversity and a +5% improvement in co-designability, validating the dataset's broad utility.
摘要: High-quality training datasets are crucial for the development of effective protein design models, but existing synthetic datasets often include unfavorable sequence-structure pairs, impairing generative model performance. We leverage ProteinMPNN, whose sequences are experimentally favorable as well as amenable to folding, together with structure prediction models to align high-quality synthetic structures with recoverable synthetic sequences. In that way, we create a new dataset designed specifically for training expressive, fully atomistic protein generators. By retraining La-Proteína, which models discrete residue type and side chain structure in a continuous latent space, on this dataset, we achieve new state-of-the-art results, with improvements of +54% in structural diversity and +27% in co-designability. To validate the broad utility of our approach, we further introduce Proteína-Atomística, a unified flow-based framework that jointly learns the distribution of protein backbone structure, discrete sequences, and atomistic side chains without latent variables. We again find that training on our new sequence-structure data dramatically boosts benchmark performance, improving Proteína-Atomística’s structural diversity by +73% and co-designability by +5%. Our work highlights the critical importance of aligned sequence-structure data for training high-performance de novo protein design models. All data will be publicly released.