
Paper List
-
Evolutionarily Stable Stackelberg Equilibrium
通过要求追随者策略对突变入侵具有鲁棒性,弥合了斯塔克尔伯格领导力模型与演化稳定性之间的鸿沟。
-
Recovering Sparse Neural Connectivity from Partial Measurements: A Covariance-Based Approach with Granger-Causality Refinement
通过跨多个实验会话累积协方差统计,实现从部分记录到完整神经连接性的重建。
-
Atomic Trajectory Modeling with State Space Models for Biomolecular Dynamics
ATMOS通过提供一个基于SSM的高效框架,用于生物分子的原子级轨迹生成,弥合了计算昂贵的MD模拟与时间受限的深度生成模型之间的差距。
-
Slow evolution towards generalism in a model of variable dietary range
通过证明是种群统计噪声(而非确定性动力学)驱动了模式形成和泛化食性的演化,解决了间接竞争下物种形成的悖论。
-
Grounded Multimodal Retrieval-Augmented Drafting of Radiology Impressions Using Case-Based Similarity Search
通过将印象草稿基于检索到的历史病例,并采用明确引用和基于置信度的拒绝机制,解决放射学报告生成中的幻觉问题。
-
Unified Policy–Value Decomposition for Rapid Adaptation
通过双线性分解在策略和价值函数之间共享低维目标嵌入,实现对新颖任务的零样本适应。
-
Mathematical Modeling of Cancer–Bacterial Therapy: Analysis and Numerical Simulation via Physics-Informed Neural Networks
提供了一个严格的、无网格的PINN框架,用于模拟和分析细菌癌症疗法中复杂的、空间异质的相互作用。
-
Sample-Efficient Adaptation of Drug-Response Models to Patient Tumors under Strong Biological Domain Shift
通过从无标记分子谱中学习可迁移表征,利用最少的临床数据实现患者药物反应的有效预测。
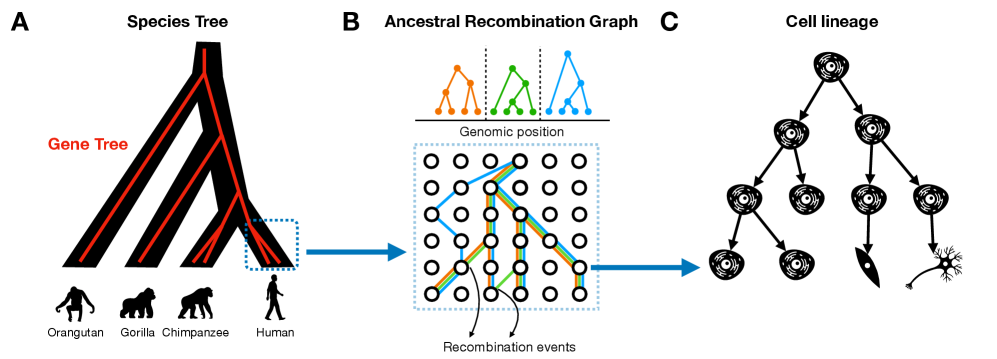
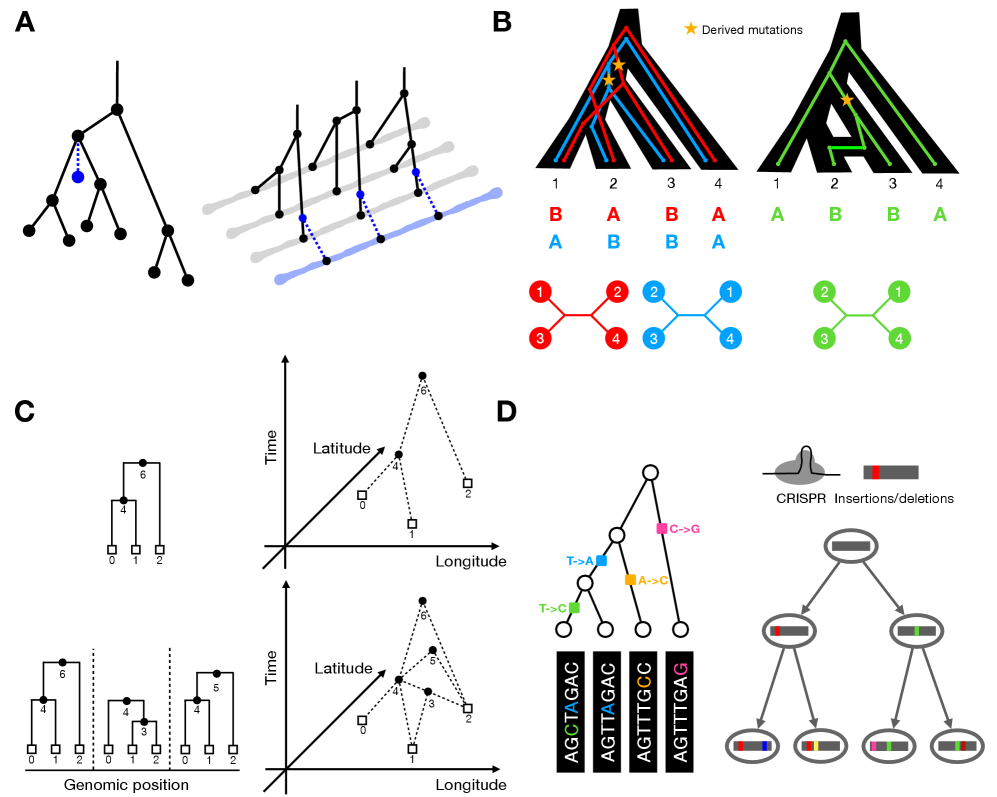
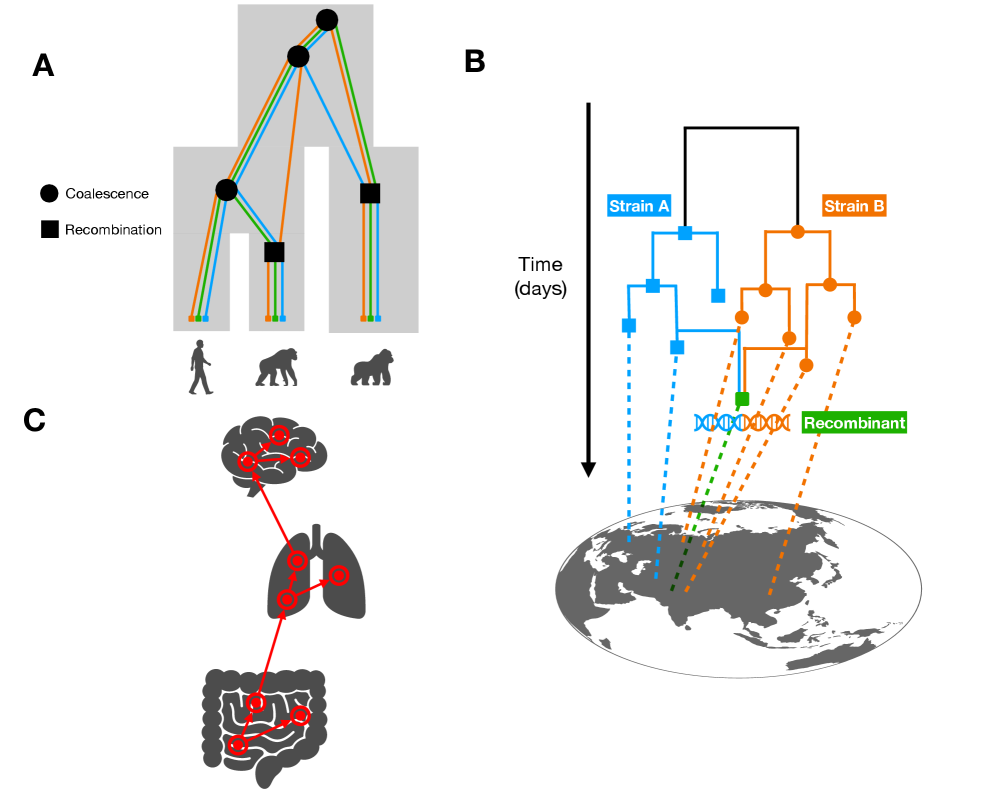
Tree Thinking in the Genomic Era: Unifying Models Across Cells, Populations, and Species
Stanford University | University of Oxford | University of California, Berkeley | Peking University | Guangzhou Medical University
30秒速读
IN SHORT: This paper addresses the fragmentation of tree-based inference methods across biological scales by identifying shared algorithmic principles and statistical challenges in phylogenetics, population genetics, and cell lineage tracing.
核心创新
- Methodology Identifies deep conceptual parallels between phylogenetic placement algorithms and ARG threading methods, demonstrating how phylogenetic placement generalizes to ARG reconstruction.
- Biology Shows that quartet-based network methods in phylogenetics and ABBA-BABA statistics in population genetics capture the same underlying signal of gene flow through asymmetric genealogical relationships.
- Methodology Demonstrates how ARG-based migration inference methods (e.g., GAIA, spacetrees) extend classical phylogeographic approaches by leveraging the full sequence of locally correlated genealogies along the genome.
主要结论
- Tree-based models provide a unified framework for ancestry inference across biological scales, with ARGs representing ~2.48 million SARS-CoV-2 genomes demonstrating pandemic-scale feasibility.
- Methodological parallels exist across domains: phylogenetic placement algorithms share core logic with ARG threading, and quartet-based methods in phylogenetics mirror ABBA-BABA statistics in population genetics for detecting gene flow.
- Current ARG inference algorithms remain constrained by simplifying assumptions (neutrality, panmixia, constant population size) and face challenges in uncertainty quantification, particularly for non-model species or limited sample sizes.
摘要: The ongoing explosion of genome sequence data is transforming how we reconstruct and understand the histories of biological systems. Across biological scales–from individual cells to populations and species–trees-based models provide a common framework for representing ancestry. Once limited to species phylogenetics, “tree thinking” now extends deeply to population genomics and cell biology, revealing the genealogical structure of genetic and phenotypic variation within and across organisms. Recently, there have been great methodological and computational advances on tree-based methods, including methods for inferring ancestral recombination graphs in populations, phylogenetic frameworks for comparative genomics, and lineage-tracing techniques in developmental and cancer biology. Despite differences in data types and biological contexts, these approaches share core statistical and algorithmic challenges: efficiently inferring branching histories from genomic information, integrating temporal and spatial signals, and connecting genealogical structures to evolutionary and functional processes. Recognizing these shared foundations opens opportunities for cross-fertilization between fields that are traditionally studied in isolation. By examining how tree-based methods are applied across cellular, population, and species scales, we identify the conceptual parallels that unite them and the distinct challenges that each domain presents. These comparisons offer new perspectives that can inform algorithmic innovations and lead to more powerful inference strategies across the full spectrum of biological systems.