
Paper List
-
Evolutionarily Stable Stackelberg Equilibrium
通过要求追随者策略对突变入侵具有鲁棒性,弥合了斯塔克尔伯格领导力模型与演化稳定性之间的鸿沟。
-
Recovering Sparse Neural Connectivity from Partial Measurements: A Covariance-Based Approach with Granger-Causality Refinement
通过跨多个实验会话累积协方差统计,实现从部分记录到完整神经连接性的重建。
-
Atomic Trajectory Modeling with State Space Models for Biomolecular Dynamics
ATMOS通过提供一个基于SSM的高效框架,用于生物分子的原子级轨迹生成,弥合了计算昂贵的MD模拟与时间受限的深度生成模型之间的差距。
-
Slow evolution towards generalism in a model of variable dietary range
通过证明是种群统计噪声(而非确定性动力学)驱动了模式形成和泛化食性的演化,解决了间接竞争下物种形成的悖论。
-
Grounded Multimodal Retrieval-Augmented Drafting of Radiology Impressions Using Case-Based Similarity Search
通过将印象草稿基于检索到的历史病例,并采用明确引用和基于置信度的拒绝机制,解决放射学报告生成中的幻觉问题。
-
Unified Policy–Value Decomposition for Rapid Adaptation
通过双线性分解在策略和价值函数之间共享低维目标嵌入,实现对新颖任务的零样本适应。
-
Mathematical Modeling of Cancer–Bacterial Therapy: Analysis and Numerical Simulation via Physics-Informed Neural Networks
提供了一个严格的、无网格的PINN框架,用于模拟和分析细菌癌症疗法中复杂的、空间异质的相互作用。
-
Sample-Efficient Adaptation of Drug-Response Models to Patient Tumors under Strong Biological Domain Shift
通过从无标记分子谱中学习可迁移表征,利用最少的临床数据实现患者药物反应的有效预测。
Incorporating indel channels into average-case analysis of seed-chain-extend
Carnegie Mellon University, Pittsburgh, PA, USA
30秒速读
IN SHORT: This paper addresses the core pain point of bridging the theoretical gap for the widely used seed-chain-extend heuristic by providing the first rigorous average-case analysis that accounts for insertions and deletions (indels), not just substitutions.
核心创新
- Methodology Introduces a generalized definition of 'recoverability' and a 'homologous path' to mathematically model the correct alignment under indel mutation channels, moving beyond the simpler 'homologous diagonal' used for substitutions only.
- Theory Develops new mathematical machinery to handle the dependence structure of neighboring anchors and the existence of 'clipping anchors' (partially correct anchors), which are unique challenges introduced by indels.
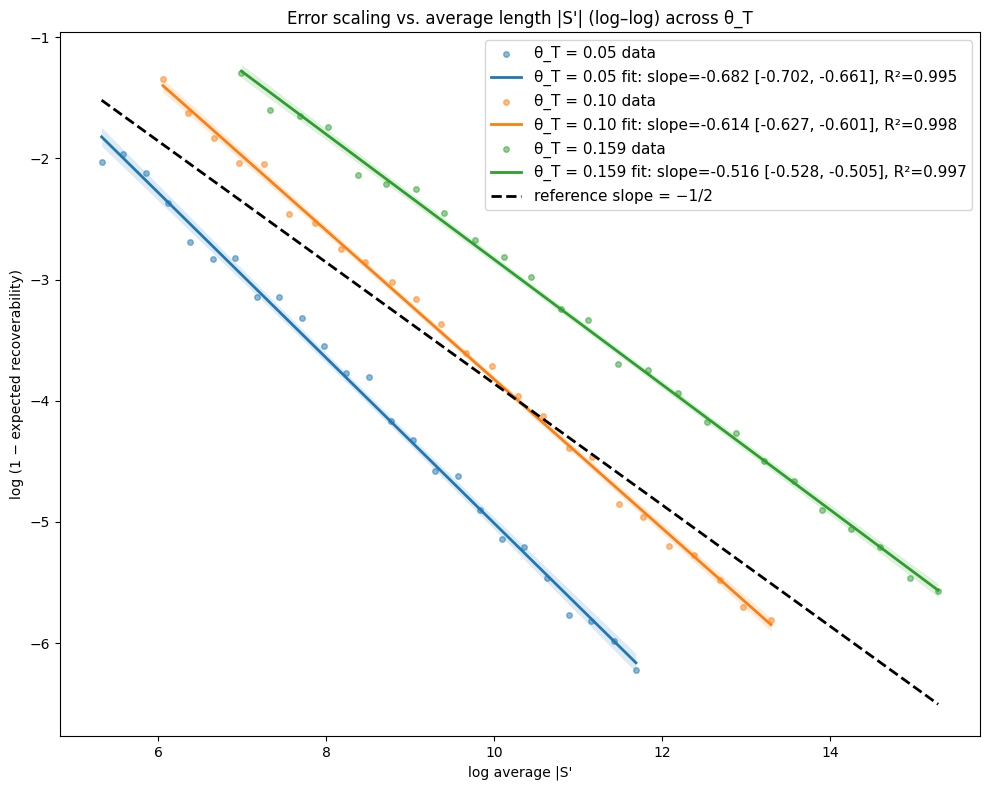
- Theory Proves that under a total mutation rate θ_T < 0.159, optimal linear-gap cost chaining achieves an expected recoverability of ≥ 1 - O(1/√m), generalizing the prior substitution-only result to a biologically realistic model.
主要结论
- The expected recoverability of an optimal chain under linear-gap cost chaining is ≥ 1 - O(1/√m) when the total mutation rate θ_T (sum of substitution, insertion, deletion rates) is less than 0.159.
- The expected runtime of the algorithm is O(m n^(3.15·θ_T) log n). For example, at a θ_T of 0.05 (similar to human-chimp divergence), the exponent is ~1.12, leading to near-linear scaling.
- The analysis successfully bridges theory and practice by extending the proof framework to handle indels, justifying the heuristic's empirical effectiveness on real genomic data which contains indels.
摘要: Given a sequence s1 of n letters drawn i.i.d. from an alphabet of size σ and a mutated substring s2 of length m<n, we often want to recover the mutation history that generated s2 from s1. Modern sequence aligners are widely used for this task, and many employ the seed-chain-extend heuristic with k-mer seeds. Previously, Shaw and Yu showed that optimal linear-gap cost chaining can produce a chain with 1−O(1/m) recoverability, the proportion of the mutation history that is recovered, in O(mn^(2.43θ) log n) expected time, where θ<0.206 is the mutation rate under a substitution-only channel and s1 is assumed to be uniformly random. However, a gap remains between theory and practice, since real genomic data includes insertions and deletions (indels), and yet seed-chain-extend remains effective. In this paper, we generalize those prior results by introducing mathematical machinery to deal with the two new obstacles introduced by indel channels: the dependence of neighboring anchors and the presence of anchors that are only partially correct. We are thus able to prove that the expected recoverability of an optimal chain is ≥1−O(1/√m) and the expected runtime is O(mn^(3.15·θ_T) log n), when the total mutation rate given by the sum of the substitution, insertion, and deletion mutation rates (θ_T = θ_i + θ_d + θ_s) is less than 0.159.