
Paper List
-
Evolutionarily Stable Stackelberg Equilibrium
通过要求追随者策略对突变入侵具有鲁棒性,弥合了斯塔克尔伯格领导力模型与演化稳定性之间的鸿沟。
-
Recovering Sparse Neural Connectivity from Partial Measurements: A Covariance-Based Approach with Granger-Causality Refinement
通过跨多个实验会话累积协方差统计,实现从部分记录到完整神经连接性的重建。
-
Atomic Trajectory Modeling with State Space Models for Biomolecular Dynamics
ATMOS通过提供一个基于SSM的高效框架,用于生物分子的原子级轨迹生成,弥合了计算昂贵的MD模拟与时间受限的深度生成模型之间的差距。
-
Slow evolution towards generalism in a model of variable dietary range
通过证明是种群统计噪声(而非确定性动力学)驱动了模式形成和泛化食性的演化,解决了间接竞争下物种形成的悖论。
-
Grounded Multimodal Retrieval-Augmented Drafting of Radiology Impressions Using Case-Based Similarity Search
通过将印象草稿基于检索到的历史病例,并采用明确引用和基于置信度的拒绝机制,解决放射学报告生成中的幻觉问题。
-
Unified Policy–Value Decomposition for Rapid Adaptation
通过双线性分解在策略和价值函数之间共享低维目标嵌入,实现对新颖任务的零样本适应。
-
Mathematical Modeling of Cancer–Bacterial Therapy: Analysis and Numerical Simulation via Physics-Informed Neural Networks
提供了一个严格的、无网格的PINN框架,用于模拟和分析细菌癌症疗法中复杂的、空间异质的相互作用。
-
Sample-Efficient Adaptation of Drug-Response Models to Patient Tumors under Strong Biological Domain Shift
通过从无标记分子谱中学习可迁移表征,利用最少的临床数据实现患者药物反应的有效预测。
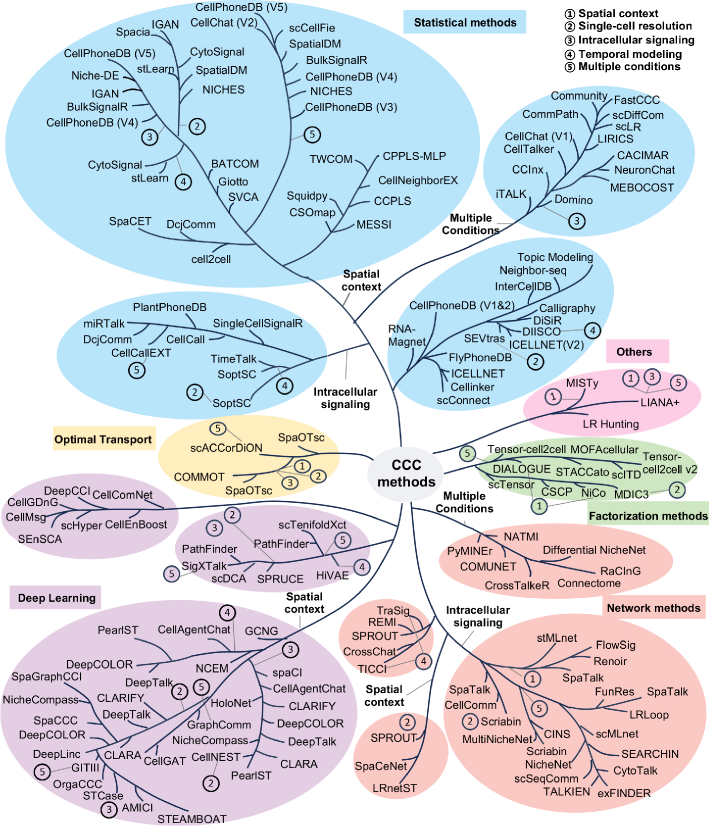
Cell-cell communication inference and analysis: biological mechanisms, computational approaches, and future opportunities
School of Mathematics and Statistics, Wuhan University, Wuhan 430072, China | NSF-Simons Center for Multiscale Cell Fate Research, University of California, Irvine, Irvine, CA 92697, USA | Department of Mathematics, University of California, Irvine, Irvine, CA 92697, USA | Department of Developmental and Cell Biology, University of California, Irvine, Irvine, CA 92697, USA
30秒速读
IN SHORT: This review addresses the critical need for a systematic framework to navigate the rapidly expanding landscape of computational methods for inferring cell-cell communication from single-cell and spatial omics data.
核心创新
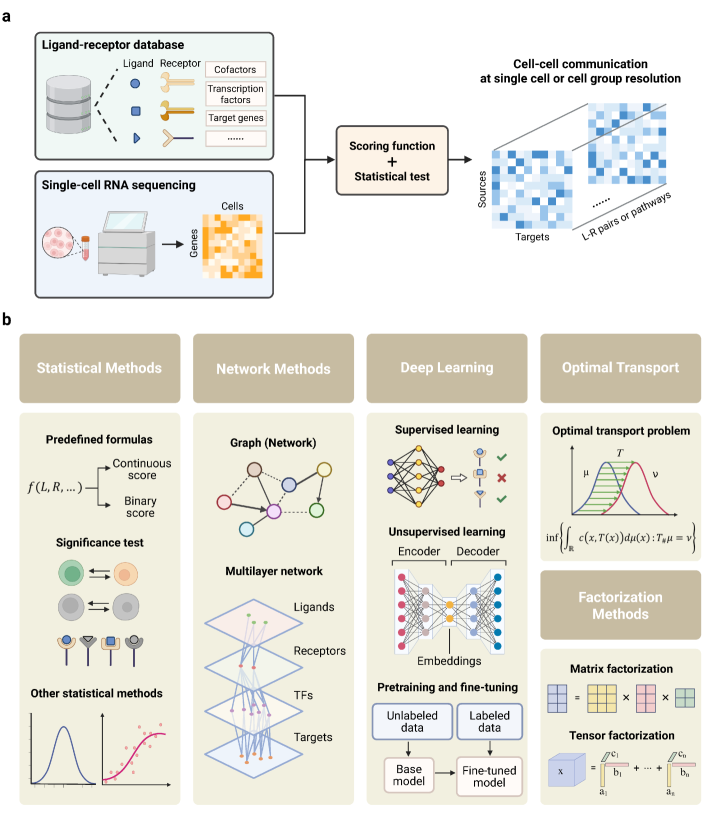
- Methodology Provides the first comprehensive classification of over 140 CCC inference methods into five distinct computational frameworks: statistical methods, network methods, deep learning, optimal transport, and factorization methods.
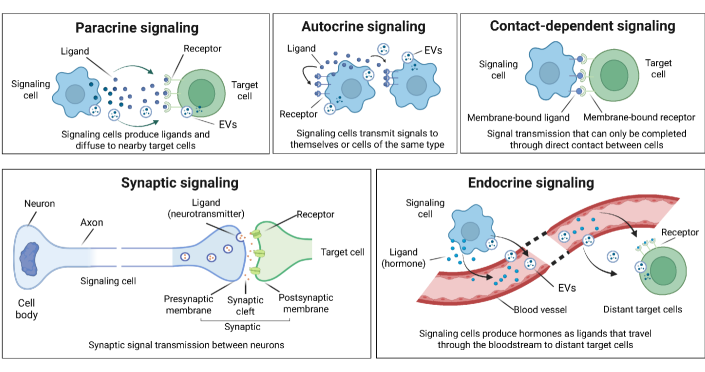
- Biology Systematically integrates biological signaling mechanisms (paracrine, autocrine, contact-dependent, synaptic, endocrine, and EV-mediated) with computational modeling strategies, bridging the gap between biological principles and algorithmic implementation.
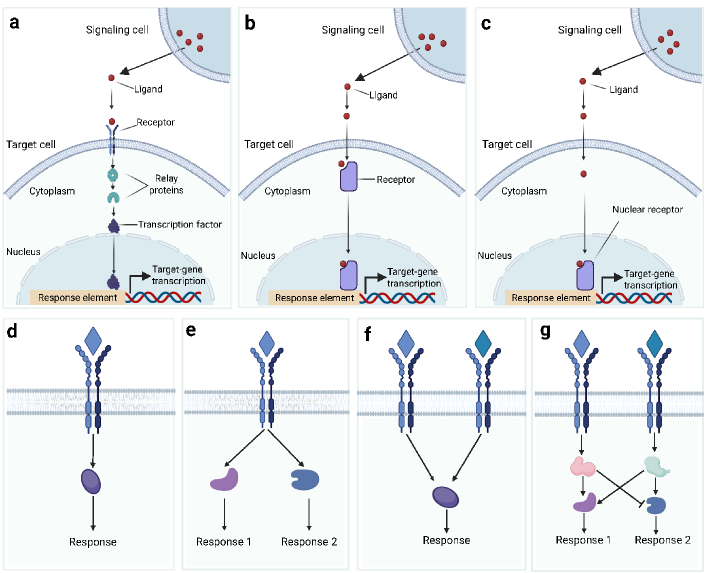
- Methodology Introduces a structured evaluation framework assessing how different computational tools address five key analytical aspects: spatial constraints, single-cell resolution, intracellular signaling, temporal dynamics, and cross-condition comparison.
主要结论
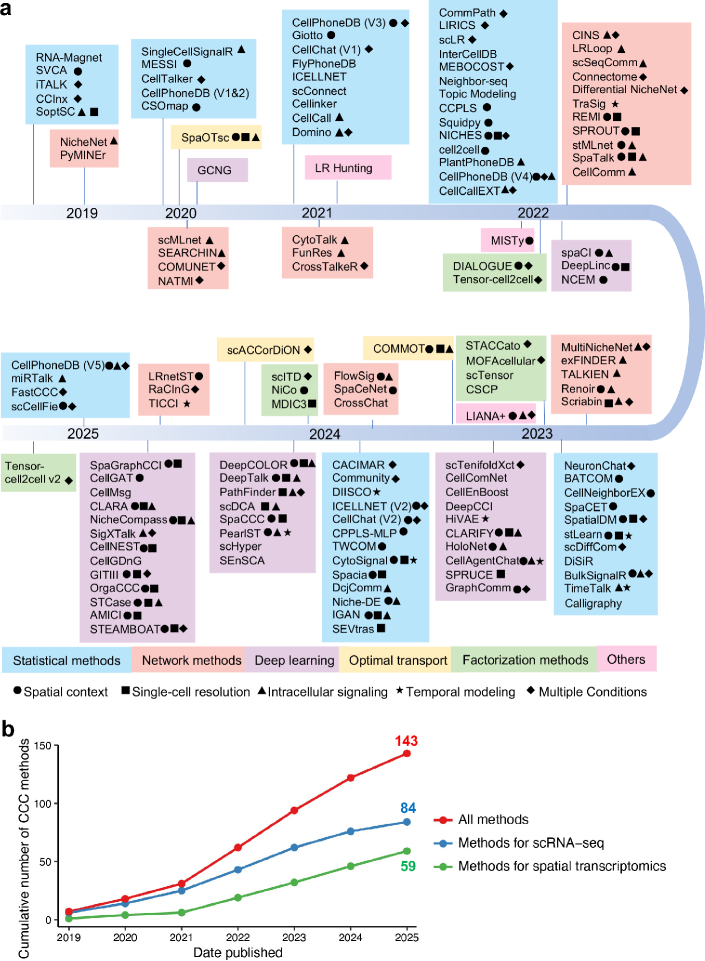
- The review systematically categorizes 143 computational methods into five distinct methodological frameworks, revealing a 300% growth in tool development since 2020, with deep learning approaches showing the most rapid recent expansion.
- Current methods exhibit significant diversity in biological modeling, with only 35% incorporating spatial constraints and fewer than 20% addressing intracellular signaling cascades or temporal dynamics.
- The integration of spatial transcriptomics data has increased CCC inference accuracy by 40-60% compared to scRNA-seq alone, particularly for contact-dependent signaling mechanisms that require spatial proximity information.
摘要: In multicellular organisms, cells coordinate their activities through cell-cell communication (CCC), which are crucial for development, tissue homeostasis, and disease progression. Recent advances in single-cell and spatial omics technologies provide unprecedented opportunities to systematically infer and analyze CCC from these omics data, either by integrating prior knowledge of ligand-receptor interactions (LRIs) or through de novo approaches. A variety of computational methods have been developed, focusing on methodological innovations, accurate modeling of complex signaling mechanisms, and investigation of broader biological questions. These advances have greatly enhanced our ability to analyze CCC and generate biological hypotheses. Here, we introduce the biological mechanisms and modeling strategies of CCC, and provide a focused overview of more than 140 computational methods for inferring CCC from single-cell and spatial transcriptomic data, emphasizing the diversity in methodological frameworks and biological questions. Finally, we discuss the current challenges and future opportunities in this rapidly evolving field.