
Paper List
-
Ill-Conditioning in Dictionary-Based Dynamic-Equation Learning: A Systems Biology Case Study
This paper addresses the critical challenge of numerical ill-conditioning and multicollinearity in library-based sparse regression methods (e.g., SIND...
-
Hybrid eTFCE–GRF: Exact Cluster-Size Retrieval with Analytical pp-Values for Voxel-Based Morphometry
This paper addresses the computational bottleneck in voxel-based neuroimaging analysis by providing a method that delivers exact cluster-size retrieva...
-
abx_amr_simulator: A simulation environment for antibiotic prescribing policy optimization under antimicrobial resistance
This paper addresses the critical challenge of quantitatively evaluating antibiotic prescribing policies under realistic uncertainty and partial obser...
-
PesTwin: a biology-informed Digital Twin for enabling precision farming
This paper addresses the critical bottleneck in precision agriculture: the inability to accurately forecast pest outbreaks in real-time, leading to su...
-
Equivariant Asynchronous Diffusion: An Adaptive Denoising Schedule for Accelerated Molecular Conformation Generation
This paper addresses the core challenge of generating physically plausible 3D molecular structures by bridging the gap between autoregressive methods ...
-
Omics Data Discovery Agents
This paper addresses the core challenge of making published omics data computationally reusable by automating the extraction, quantification, and inte...
-
Single-cell directional sensing at ultra-low chemoattractant concentrations from extreme first-passage events
This work addresses the core challenge of how a cell can rapidly and accurately determine the direction of a chemoattractant source when the signal is...
-
SDSR: A Spectral Divide-and-Conquer Approach for Species Tree Reconstruction
This paper addresses the computational bottleneck in reconstructing species trees from thousands of species and multiple genes by introducing a scalab...
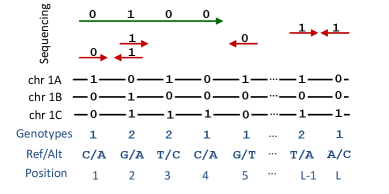
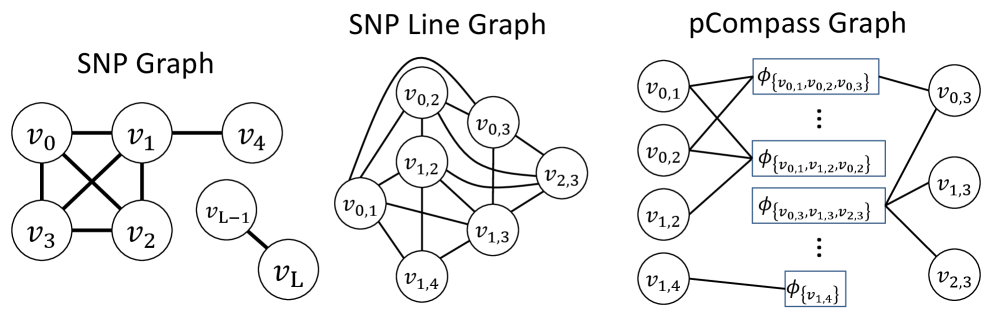
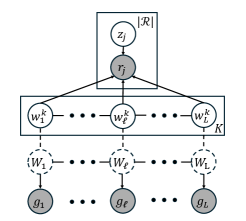
pHapCompass: Probabilistic Assembly and Uncertainty Quantification of Polyploid Haplotype Phase
School of Computing, University of Connecticut | Department of Entomology and Plant Pathology, University of Tennessee | Institute for Systems Genomics, University of Connecticut
30秒速读
IN SHORT: This paper addresses the core challenge of accurately assembling polyploid haplotypes from sequencing data, where read assignment ambiguity and an exponential search space of possible phasings have hindered reliable reconstruction and uncertainty quantification.
核心创新
- Methodology Introduces pHapCompass, the first probabilistic haplotype assembler for diploid and polyploid genomes that explicitly models read assignment ambiguity to compute a distribution over haplotype phasings, enabling formal uncertainty quantification.
- Methodology Develops two distinct graph-theoretic algorithms: pHapCompass-short (a Markov random field for high-coverage short reads) and pHapCompass-long (a hierarchical mixture model for low-coverage long reads), both designed to scale with genomic complexity.
- Methodology Creates the first computational workflow for simulating realistic auto- and allopolyploid genomes and sequencing data, addressing a critical gap in benchmarking tools that previously relied on oversimplified synthetic genomes.
主要结论
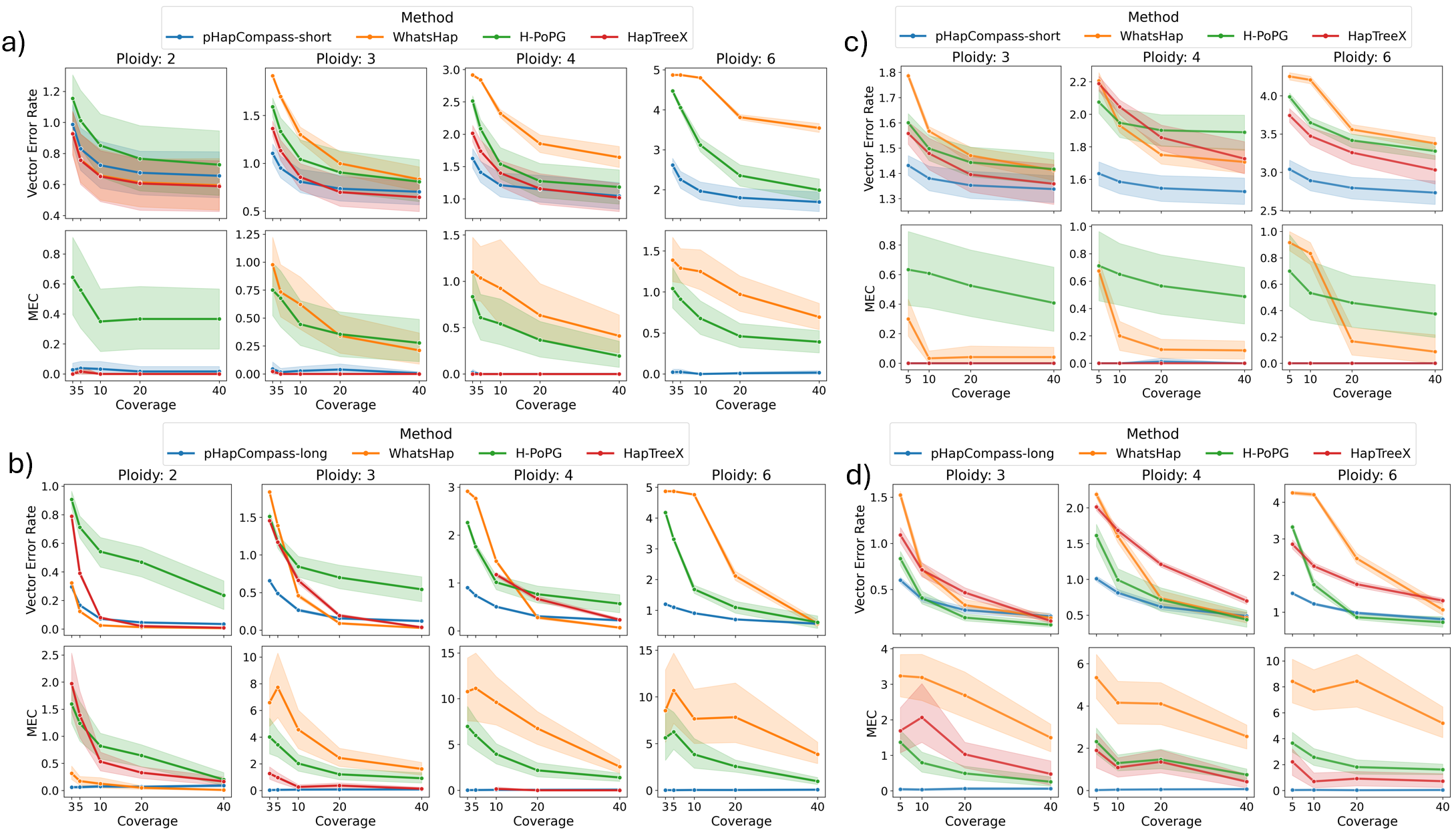
- pHapCompass demonstrates competitive performance against existing assemblers across varying ploidy levels, coverage depths, and mutation rates, while uniquely providing accurate quantification of phase uncertainty.
- The developed simulation workflow generates more realistic benchmarking datasets, revealing that prior methods often overestimate performance on simplistic synthetic genomes.
- The framework successfully assembled an allo-octoploid strawberry chromosome, showcasing practical applicability to complex, real-world polyploid genomes.
摘要: Computing haplotypes from sequencing data, i.e. haplotype assembly, is an important component of foundational molecular and population genetics problems, including interpreting the effects of genetic variation on complex traits and reconstructing genealogical relationships. Assembling the haplotypes of polyploid genomes remains a significant challenge due to the exponential search space of haplotype phasings and read assignment ambiguity; the latter challenge is particularly difficult for polyploid haplotype assemblers since the information contained within the observed sequence reads is often insufficient for unambiguous haplotype assignment in polyploid genomes. We present pHapCompass, probabilistic haplotype assembly algorithms for diploid and polyploid genomes that explicitly model and propagate read assignment ambiguity to compute a distribution over polyploid haplotype phasings. We develop graph theoretic algorithms to enable statistical inference and uncertainty quantification despite an exponential space of possible phasings. Since prior work evaluates polyploid haplotype assembly on synthetic genomes that do not reflect the realistic genomic complexity of polyploidy organisms, we develop a computational workflow for simulating genomes and DNA-seq for auto- and allopolyploids. Additionally, we generalize the vector error rate and minimum error correction evaluation criteria for partially phased haplotypes. Benchmarking of pHapCompass and several existing polyploid haplotype assemblers shows that pHapCompass yields competitive performance across varying genomic complexities and polyploid structures while retaining an accurate quantification of phase uncertainty. The source code for pHapCompass, simulation scripts, and datasets are freely available at https://github.com/bayesomicslab/pHapCompass.