
Paper List
-
SpikGPT: A High-Accuracy and Interpretable Spiking Attention Framework for Single-Cell Annotation
This paper addresses the core challenge of robust single-cell annotation across heterogeneous datasets with batch effects and the critical need to ide...
-
Unlocking hidden biomolecular conformational landscapes in diffusion models at inference time
This paper addresses the core challenge of efficiently and accurately sampling the conformational landscape of biomolecules from diffusion-based struc...
-
Personalized optimization of pediatric HD-tDCS for dose consistency and target engagement
This paper addresses the critical limitation of one-size-fits-all HD-tDCS protocols in pediatric populations by developing a personalized optimization...
-
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein con...
-
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
This paper addresses the core pain point of low sequence-structure alignment in existing synthetic datasets (e.g., AFDB), which severely limits the pe...
-
MoRSAIK: Sequence Motif Reactor Simulation, Analysis and Inference Kit in Python
This work addresses the computational bottleneck in simulating prebiotic RNA reactor dynamics by developing a Python package that tracks sequence moti...
-
On the Approximation of Phylogenetic Distance Functions by Artificial Neural Networks
This paper addresses the core challenge of developing computationally efficient and scalable neural network architectures that can learn accurate phyl...
-
EcoCast: A Spatio-Temporal Model for Continual Biodiversity and Climate Risk Forecasting
This paper addresses the critical bottleneck in conservation: the lack of timely, high-resolution, near-term forecasts of species distribution shifts ...
GOPHER: Optimization-based Phenotype Randomization for Genome-Wide Association Studies with Differential Privacy
Department of Biomedical Informatics & Data Science, Yale School of Medicine | Department of Technology and Operations Management, Harvard Business School | Department of Computer Science, Yale University
30秒速读
IN SHORT: This paper addresses the core challenge of balancing rigorous privacy protection with data utility when releasing full GWAS summary statistics, overcoming the limitations of prior methods that either add excessive noise or restrict output to a small subset of results.
核心创新
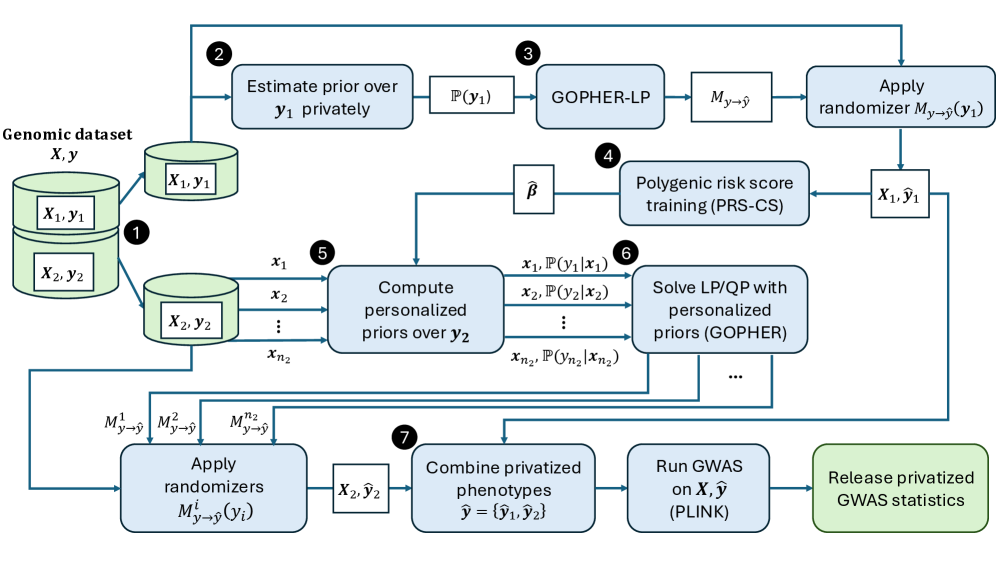
- Methodology Introduces an optimization-based phenotype randomization mechanism (GOPHER-LP) that directly minimizes expected error in GWAS statistics, formulated as a linear programming problem to enhance utility beyond baseline methods like randomized response.
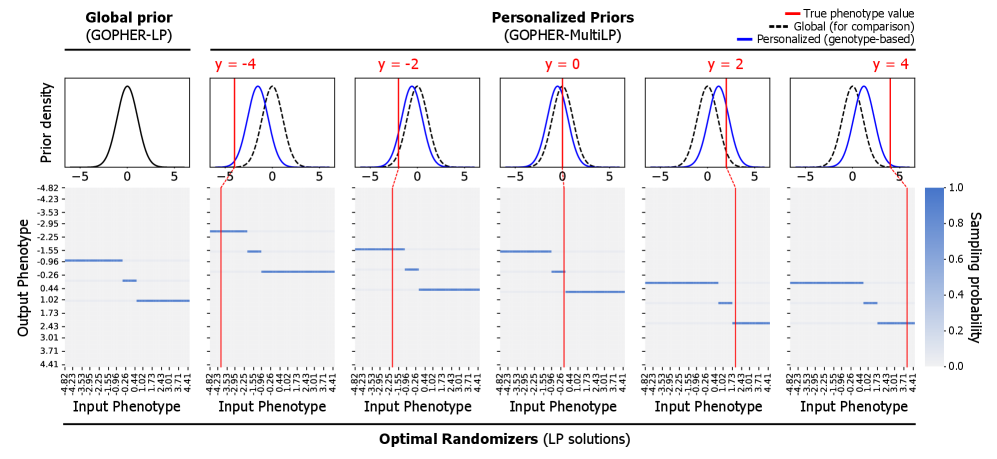
- Methodology Proposes GOPHER-MultiLP, which incorporates personalized priors derived from predictive models (e.g., polygenic risk scores) trained on a held-out subset, enabling sample-specific optimization that leverages genotype information to further reduce noise.
- Theory Adopts and extends the concept of phenotypic differential privacy (analogous to label DP), focusing protection on sensitive phenotypes while treating genotypes as public, providing a practical middle ground between full DP and unrestricted release.
主要结论
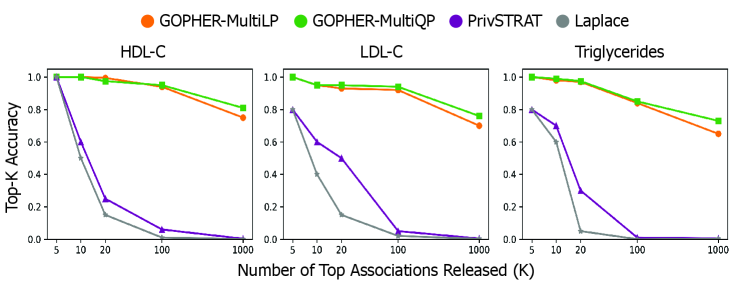
- The GOPHER framework enables the release of complete GWAS statistics (e.g., over 500,000 variants) with provable privacy guarantees, a significant scalability advance over prior methods limited to releasing only 3-5 top associations.
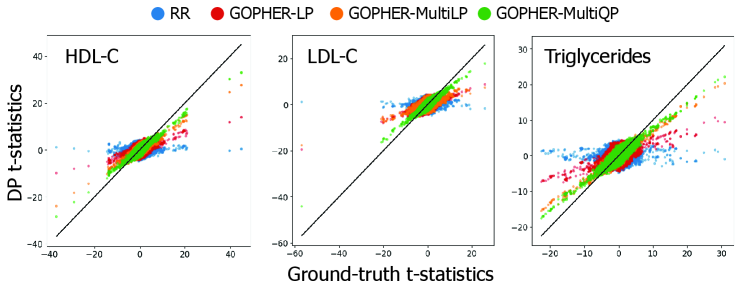
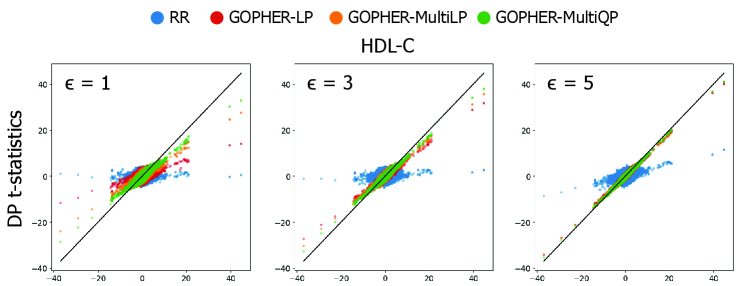
- Experiments on UK Biobank data (n=100,000) demonstrate that the mechanisms yield association statistics that accurately match non-private GWAS results while maintaining rigorous (ε, δ)-DP guarantees.
- The phenotype-randomization approach decouples the added noise from the number of genetic variants analyzed, addressing a fundamental scalability challenge not previously solved in the DP-GWAS literature.
摘要: Genome-wide association studies (GWAS) are an essential tool in biomedical research for identifying genetic factors linked to health and disease. However, publicly releasing GWAS summary statistics poses well-recognized privacy risks, including the potential to infer an individual’s participation in the study or to reveal sensitive phenotypic information (e.g., disease status). While differential privacy (DP) offers a rigorous mathematical framework for mitigating these risks, existing DP techniques for GWAS either introduce excessive noise or restrict the release to a limited set of results. In this work, we present practical DP mechanisms for releasing the complete set of genome-wide association statistics with privacy guarantees. We demonstrate the accuracy of the privacy-preserving statistics released by our mechanisms on a range of GWAS datasets from the UK Biobank, utilizing both real and simulated phenotypes. We introduce two key techniques to overcome the limitations of prior approaches: (1) an optimization-based randomization mechanism that directly minimizes the expected error in GWAS results to enhance utility, and (2) the use of personalized priors, derived from predictive models privately trained on a subset of the dataset, to enable sample-specific optimization which further reduces the amount of noise introduced by DP. Overall, our work provides practical tools for accurately releasing comprehensive GWAS results with provable protection of study participants.