
Paper List
-
SpikGPT: A High-Accuracy and Interpretable Spiking Attention Framework for Single-Cell Annotation
This paper addresses the core challenge of robust single-cell annotation across heterogeneous datasets with batch effects and the critical need to ide...
-
Unlocking hidden biomolecular conformational landscapes in diffusion models at inference time
This paper addresses the core challenge of efficiently and accurately sampling the conformational landscape of biomolecules from diffusion-based struc...
-
Personalized optimization of pediatric HD-tDCS for dose consistency and target engagement
This paper addresses the critical limitation of one-size-fits-all HD-tDCS protocols in pediatric populations by developing a personalized optimization...
-
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein con...
-
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
This paper addresses the core pain point of low sequence-structure alignment in existing synthetic datasets (e.g., AFDB), which severely limits the pe...
-
MoRSAIK: Sequence Motif Reactor Simulation, Analysis and Inference Kit in Python
This work addresses the computational bottleneck in simulating prebiotic RNA reactor dynamics by developing a Python package that tracks sequence moti...
-
On the Approximation of Phylogenetic Distance Functions by Artificial Neural Networks
This paper addresses the core challenge of developing computationally efficient and scalable neural network architectures that can learn accurate phyl...
-
EcoCast: A Spatio-Temporal Model for Continual Biodiversity and Climate Risk Forecasting
This paper addresses the critical bottleneck in conservation: the lack of timely, high-resolution, near-term forecasts of species distribution shifts ...
Contrastive Deep Learning for Variant Detection in Wastewater Genomic Sequencing
Georgia State University, Atlanta, Georgia, USA
30秒速读
IN SHORT: This paper addresses the core challenge of detecting viral variants in wastewater sequencing data without reference genomes or labeled annotations, overcoming issues of high noise, low coverage, and fragmented reads.
核心创新
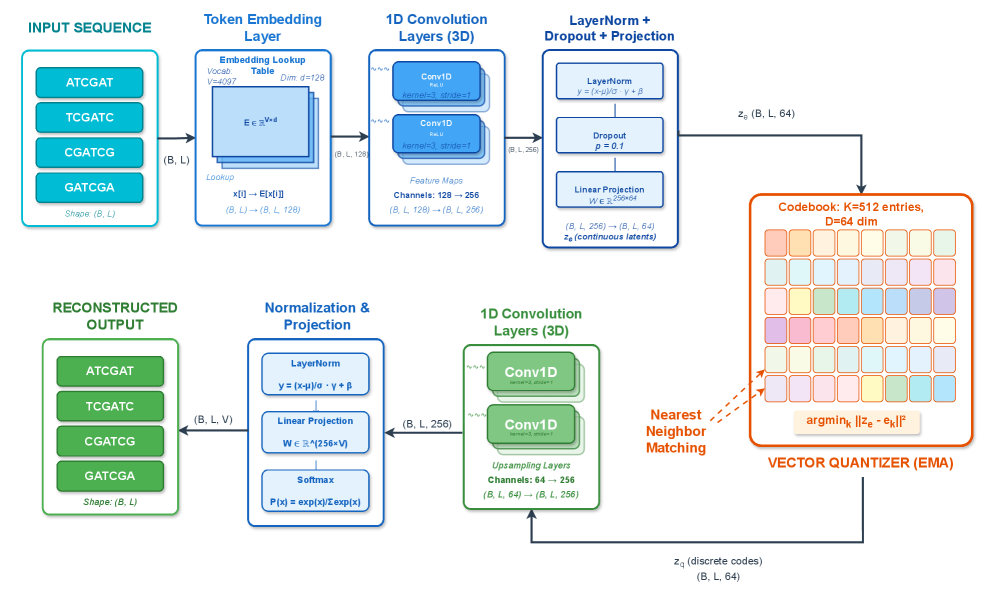
- Methodology First comprehensive application of VQ-VAE with EMA quantization to wastewater genomic surveillance, achieving 99.52% token-level reconstruction accuracy with 19.73% codebook utilization.
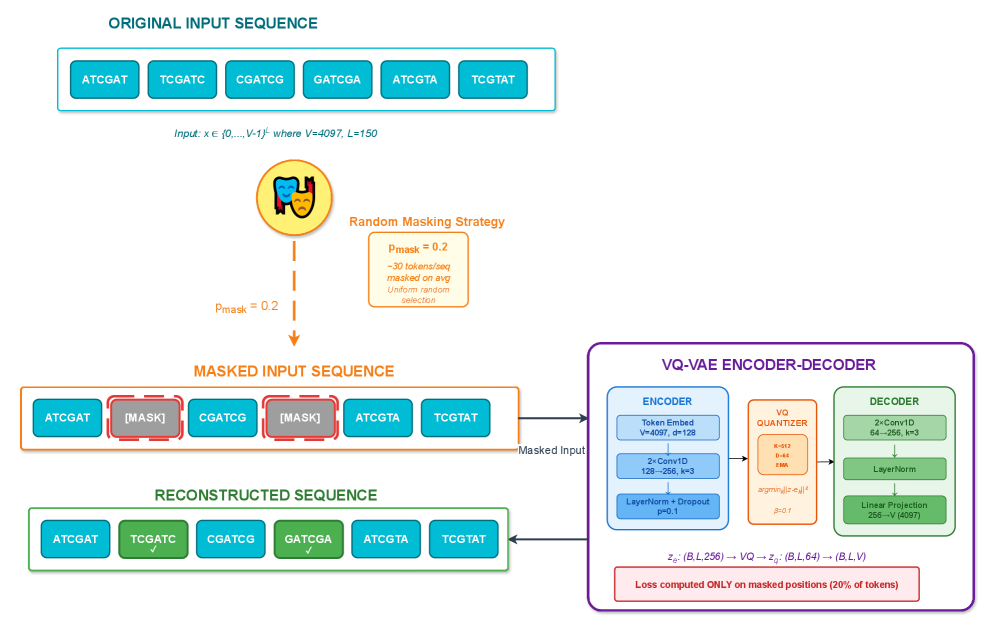
- Methodology Integration of masked reconstruction pretraining (BERT-style) maintaining ~95% accuracy under 20% token corruption, enabling robust inference with missing/low-quality data.
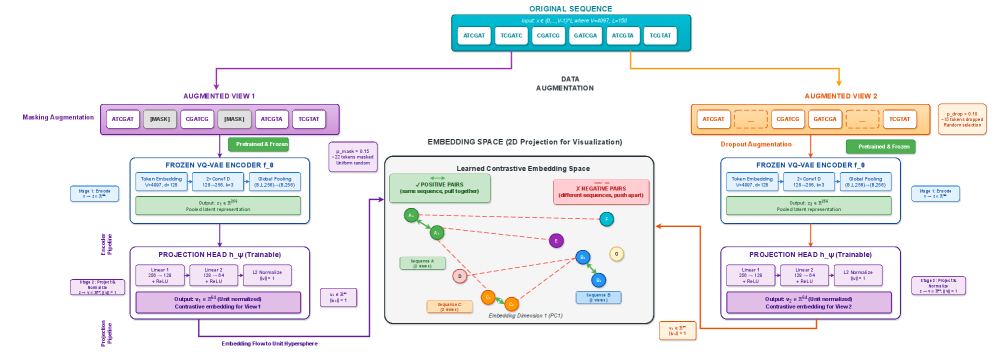
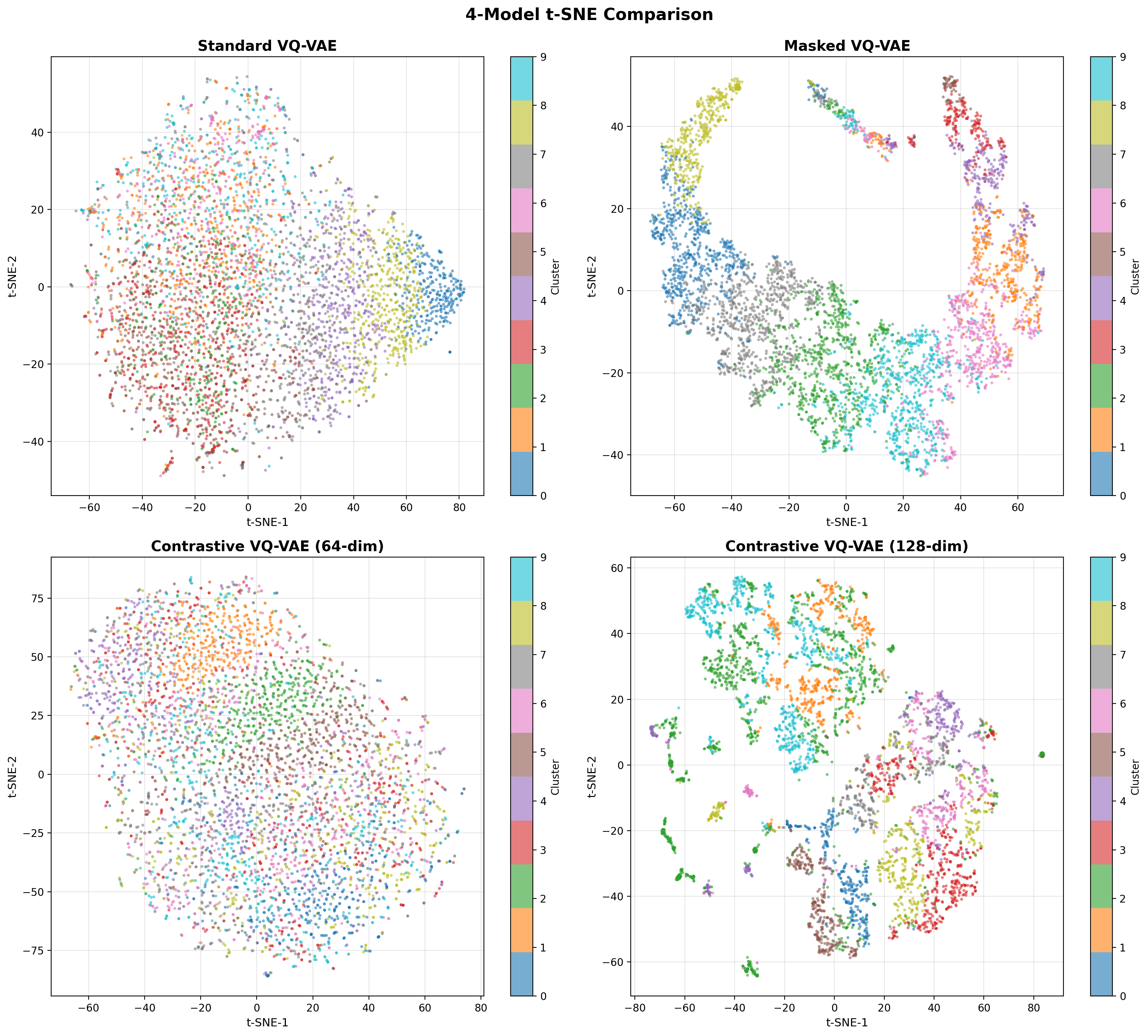
- Methodology Contrastive fine-tuning with varying embedding dimensions showing +35% (64-dim) and +42% (128-dim) Silhouette score improvements, establishing representation capacity impact on variant discrimination.
主要结论
- VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate on SARS-CoV-2 wastewater data with 100,000 reads.
- Contrastive fine-tuning improves clustering performance by +35% (0.31→0.42) with 64-dim embeddings and +42% (0.31→0.44) with 128-dim embeddings.
- The framework maintains efficient codebook utilization (19.73%, 101 of 512 codes active) while providing robust performance under data corruption.
摘要: Wastewater-based genomic surveillance has emerged as a powerful tool for population-level viral monitoring, offering comprehensive insights into circulating viral variants across entire communities. However, this approach faces significant computational challenges stemming from high sequencing noise, low viral coverage, fragmented reads, and the complete absence of labeled variant annotations. Traditional reference-based variant calling pipelines struggle with novel mutations and require extensive computational resources. We present a comprehensive framework for unsupervised viral variant detection using Vector-Quantized Variational Autoencoders (VQ-VAE) that learns discrete codebooks of genomic patterns from k-mer tokenized sequences without requiring reference genomes or variant labels. Our approach extends the base VQ-VAE architecture with masked reconstruction pretraining for robustness to missing data and contrastive learning for highly discriminative embeddings. Evaluated on SARS-CoV-2 wastewater sequencing data comprising approximately 100,000 reads, our VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate while maintaining 19.73% codebook utilization (101 of 512 codes active), demonstrating efficient discrete representation learning. Contrastive fine-tuning with different projection dimensions yields substantial clustering improvements: 64-dimensional embeddings achieve +35% Silhouette score improvement (0.31→0.42), while 128-dimensional embeddings achieve +42% improvement (0.31→0.44), clearly demonstrating the impact of embedding dimensionality on variant discrimination capability. Our reference-free framework provides a scalable, interpretable approach to genomic surveillance with direct applications to public health monitoring.