
Paper List
-
GOPHER: Optimization-based Phenotype Randomization for Genome-Wide Association Studies with Differential Privacy
This paper addresses the core challenge of balancing rigorous privacy protection with data utility when releasing full GWAS summary statistics, overco...
-
Real-time Cricket Sorting By Sex A low-cost embedded solution using YOLOv8 and Raspberry Pi
This paper addresses the critical bottleneck in industrial insect farming: the lack of automated, real-time sex sorting systems for Acheta domesticus ...
-
Training Dynamics of Learning 3D-Rotational Equivariance
This work addresses the core dilemma of whether to use computationally expensive equivariant architectures or faster symmetry-agnostic models with dat...
-
Fast and Accurate Node-Age Estimation Under Fossil Calibration Uncertainty Using the Adjusted Pairwise Likelihood
This paper addresses the dual challenge of computational inefficiency and sensitivity to fossil calibration errors in Bayesian divergence time estimat...
-
Few-shot Protein Fitness Prediction via In-context Learning and Test-time Training
This paper addresses the core challenge of accurately predicting protein fitness with only a handful of experimental observations, where data collecti...
-
scCluBench: Comprehensive Benchmarking of Clustering Algorithms for Single-Cell RNA Sequencing
This paper addresses the critical gap of fragmented and non-standardized benchmarking in single-cell RNA-seq clustering, which hinders objective compa...
-
Simulation and inference methods for non-Markovian stochastic biochemical reaction networks
This paper addresses the computational bottleneck of simulating and performing Bayesian inference for non-Markovian biochemical systems with history-d...
-
Assessment of Simulation-based Inference Methods for Stochastic Compartmental Models
This paper addresses the core challenge of performing accurate Bayesian parameter inference for stochastic epidemic models when the likelihood functio...
Contrastive Deep Learning for Variant Detection in Wastewater Genomic Sequencing
Georgia State University, Atlanta, Georgia, USA
30秒速读
IN SHORT: This paper addresses the core challenge of detecting viral variants in wastewater sequencing data without reference genomes or labeled annotations, overcoming issues of high noise, low coverage, and fragmented reads.
核心创新
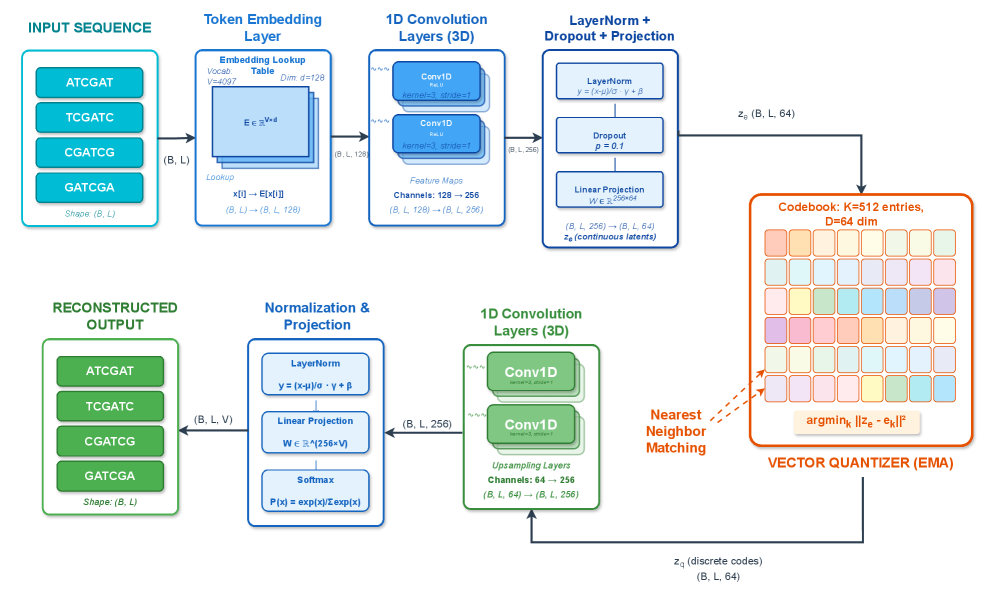
- Methodology First comprehensive application of VQ-VAE with EMA quantization to wastewater genomic surveillance, achieving 99.52% token-level reconstruction accuracy with 19.73% codebook utilization.
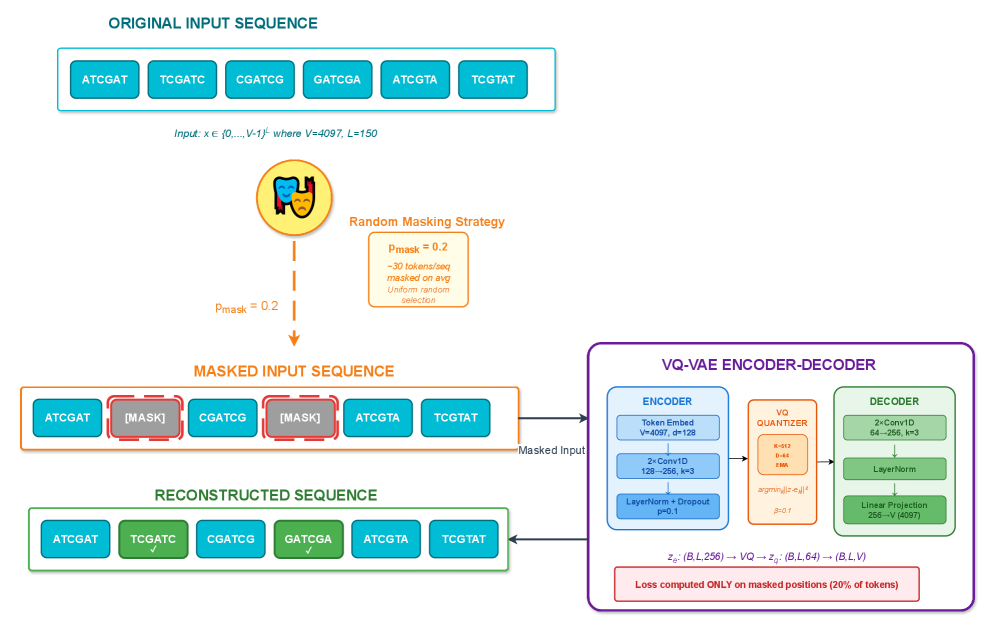
- Methodology Integration of masked reconstruction pretraining (BERT-style) maintaining ~95% accuracy under 20% token corruption, enabling robust inference with missing/low-quality data.
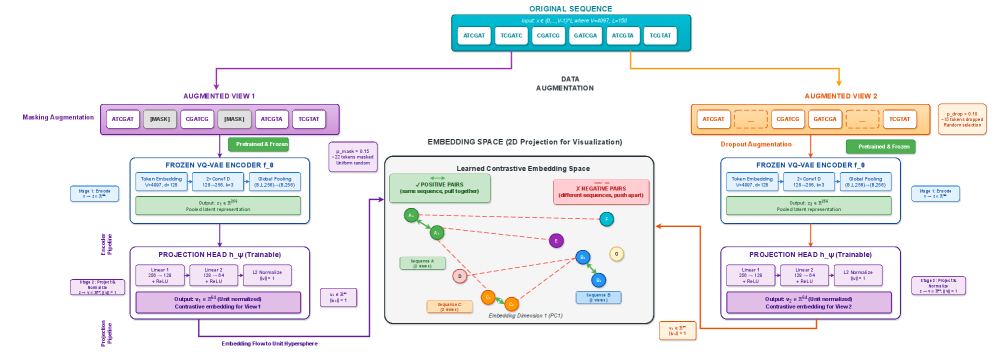
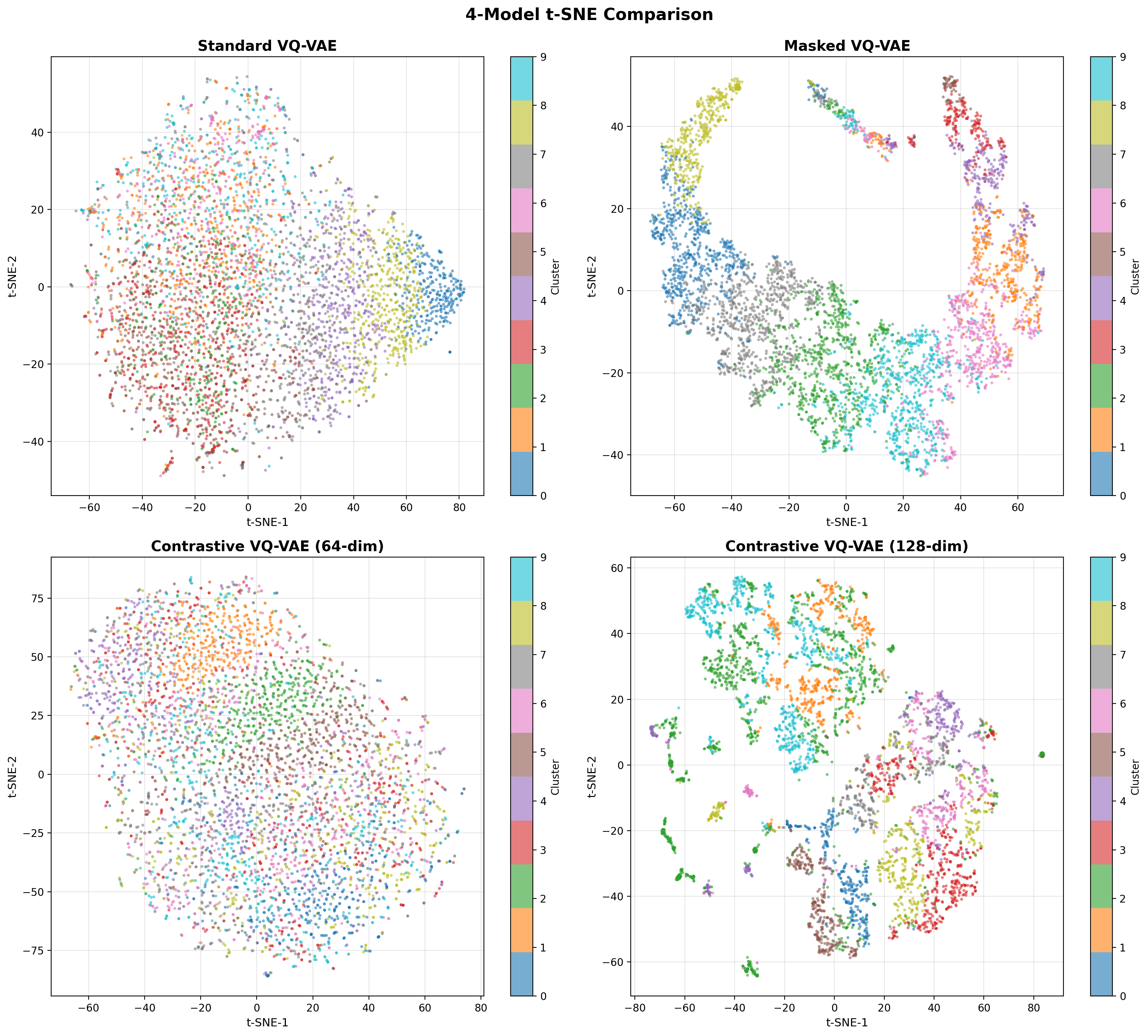
- Methodology Contrastive fine-tuning with varying embedding dimensions showing +35% (64-dim) and +42% (128-dim) Silhouette score improvements, establishing representation capacity impact on variant discrimination.
主要结论
- VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate on SARS-CoV-2 wastewater data with 100,000 reads.
- Contrastive fine-tuning improves clustering performance by +35% (0.31→0.42) with 64-dim embeddings and +42% (0.31→0.44) with 128-dim embeddings.
- The framework maintains efficient codebook utilization (19.73%, 101 of 512 codes active) while providing robust performance under data corruption.
摘要: Wastewater-based genomic surveillance has emerged as a powerful tool for population-level viral monitoring, offering comprehensive insights into circulating viral variants across entire communities. However, this approach faces significant computational challenges stemming from high sequencing noise, low viral coverage, fragmented reads, and the complete absence of labeled variant annotations. Traditional reference-based variant calling pipelines struggle with novel mutations and require extensive computational resources. We present a comprehensive framework for unsupervised viral variant detection using Vector-Quantized Variational Autoencoders (VQ-VAE) that learns discrete codebooks of genomic patterns from k-mer tokenized sequences without requiring reference genomes or variant labels. Our approach extends the base VQ-VAE architecture with masked reconstruction pretraining for robustness to missing data and contrastive learning for highly discriminative embeddings. Evaluated on SARS-CoV-2 wastewater sequencing data comprising approximately 100,000 reads, our VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate while maintaining 19.73% codebook utilization (101 of 512 codes active), demonstrating efficient discrete representation learning. Contrastive fine-tuning with different projection dimensions yields substantial clustering improvements: 64-dimensional embeddings achieve +35% Silhouette score improvement (0.31→0.42), while 128-dimensional embeddings achieve +42% improvement (0.31→0.44), clearly demonstrating the impact of embedding dimensionality on variant discrimination capability. Our reference-free framework provides a scalable, interpretable approach to genomic surveillance with direct applications to public health monitoring.