
Paper List
-
Evolutionarily Stable Stackelberg Equilibrium
通过要求追随者策略对突变入侵具有鲁棒性,弥合了斯塔克尔伯格领导力模型与演化稳定性之间的鸿沟。
-
Recovering Sparse Neural Connectivity from Partial Measurements: A Covariance-Based Approach with Granger-Causality Refinement
通过跨多个实验会话累积协方差统计,实现从部分记录到完整神经连接性的重建。
-
Atomic Trajectory Modeling with State Space Models for Biomolecular Dynamics
ATMOS通过提供一个基于SSM的高效框架,用于生物分子的原子级轨迹生成,弥合了计算昂贵的MD模拟与时间受限的深度生成模型之间的差距。
-
Slow evolution towards generalism in a model of variable dietary range
通过证明是种群统计噪声(而非确定性动力学)驱动了模式形成和泛化食性的演化,解决了间接竞争下物种形成的悖论。
-
Grounded Multimodal Retrieval-Augmented Drafting of Radiology Impressions Using Case-Based Similarity Search
通过将印象草稿基于检索到的历史病例,并采用明确引用和基于置信度的拒绝机制,解决放射学报告生成中的幻觉问题。
-
Unified Policy–Value Decomposition for Rapid Adaptation
通过双线性分解在策略和价值函数之间共享低维目标嵌入,实现对新颖任务的零样本适应。
-
Mathematical Modeling of Cancer–Bacterial Therapy: Analysis and Numerical Simulation via Physics-Informed Neural Networks
提供了一个严格的、无网格的PINN框架,用于模拟和分析细菌癌症疗法中复杂的、空间异质的相互作用。
-
Sample-Efficient Adaptation of Drug-Response Models to Patient Tumors under Strong Biological Domain Shift
通过从无标记分子谱中学习可迁移表征,利用最少的临床数据实现患者药物反应的有效预测。
Simulation and inference methods for non-Markovian stochastic biochemical reaction networks
School of Mathematical Sciences, Queensland University of Technology | Centre for Data Science, Queensland University of Technology | ARC Centre of Excellence for Mathematical Analysis of Cellular Systems (MACSYS), Queensland University of Technology
30秒速读
IN SHORT: This paper addresses the computational bottleneck of simulating and performing Bayesian inference for non-Markovian biochemical systems with history-dependent delays, which are crucial for modeling processes like gene transcription but are prohibitively expensive with existing methods.
核心创新
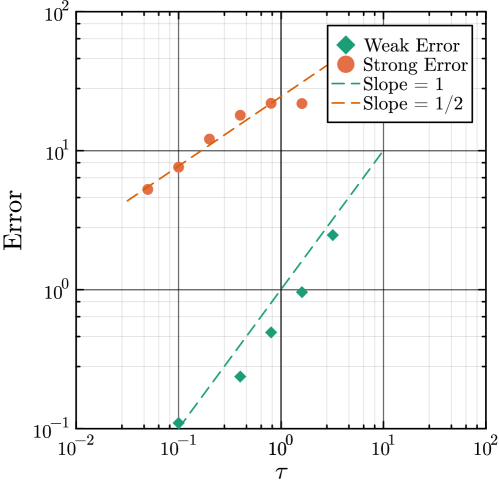
- Methodology Generalizes the next reaction method and τ-leaping to support arbitrary inter-event time distributions for non-Markovian systems, maintaining computational scalability.
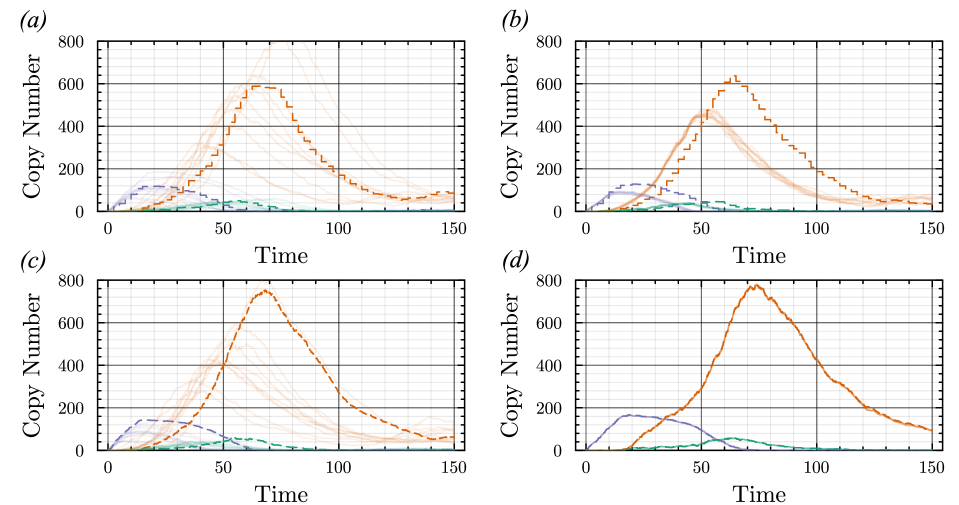
- Methodology Introduces a novel coupling scheme to generate positively correlated exact and approximate non-Markovian sample paths, a prerequisite for variance reduction techniques.
- Methodology Enables the application of multifidelity and multilevel Monte Carlo (MLMC) methods to non-Markovian systems for the first time, bridging a significant methodological gap.
主要结论
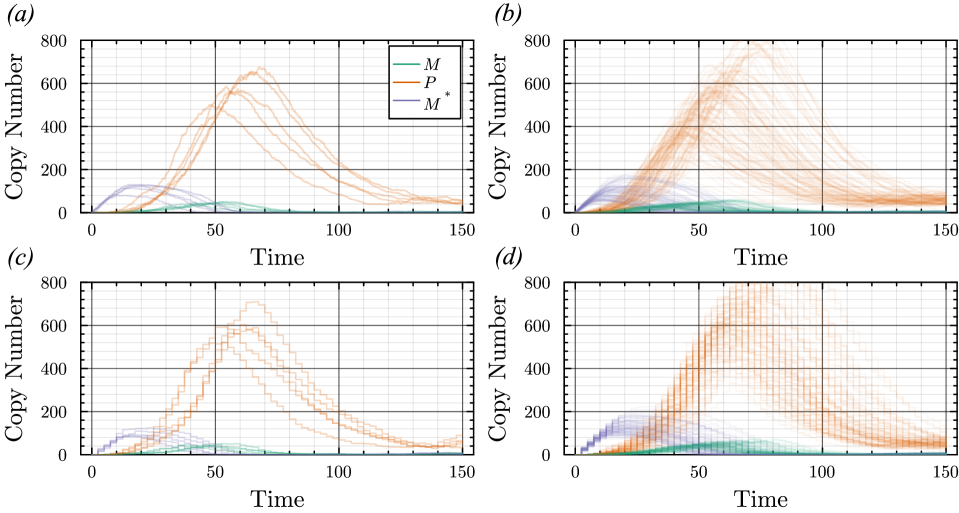
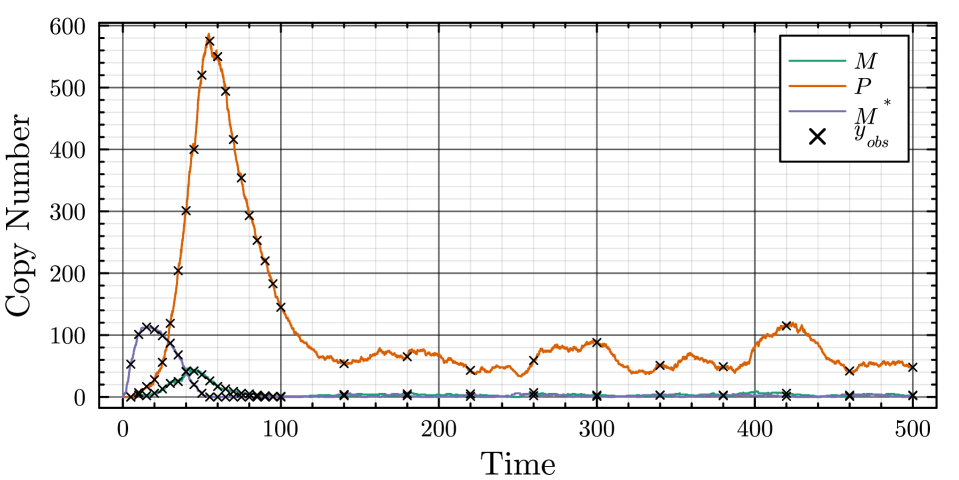
- The proposed non-Markovian simulation algorithms and coupling scheme successfully enable multifidelity inference, demonstrated on a gene regulation model with delayed auto-inhibition.
- The method achieves a computational speedup of two orders of magnitude (100x) in inference efficiency compared to standard approaches for the non-Markovian case study.
- The framework supports arbitrary delay distributions (state- and time-dependent), significantly extending the practical modeling scope beyond previous methods limited to simpler, time-only delays.
摘要: Stochastic models of biochemical reaction networks are widely used to capture intrinsic noise in cellular systems. The typical formulation of these models are based on Markov processes for which there is extensive research on efficient simulation and inference. However, there are biological processes, such as gene transcription and translation, that introduce history dependent dynamics requiring non-Markovian processes to accurately capture the stochastic dynamics of the system. This greater realism comes with additional computational challenges for simulation and parameter inference. We develop efficient stochastic simulation algorithms for well-mixed non-Markovian stochastic biochemical reaction networks with delays that depend on system state and time. Our methods generalize the next reaction method and τ-leaping method to support arbitrary inter-event time distributions while preserving computational scalability. We also introduce a coupling scheme to generate exact non-Markovian sample paths that are positively correlated to an approximate non-Markovian τ-leaping sample path. This enables substantial computational gains for Bayesian inference of model parameters though multifidelity simulation-based inference schemes. We demonstrate the effectiveness of our approach on a gene regulation model with delayed auto-inhibition, showing substantial gains in both simulation accuracy and inference efficiency of two orders of magnitude. These results extend the practical applicability of non-Markovian models in systems biology and beyond.