
Paper List
-
Ill-Conditioning in Dictionary-Based Dynamic-Equation Learning: A Systems Biology Case Study
This paper addresses the critical challenge of numerical ill-conditioning and multicollinearity in library-based sparse regression methods (e.g., SIND...
-
Hybrid eTFCE–GRF: Exact Cluster-Size Retrieval with Analytical pp-Values for Voxel-Based Morphometry
This paper addresses the computational bottleneck in voxel-based neuroimaging analysis by providing a method that delivers exact cluster-size retrieva...
-
abx_amr_simulator: A simulation environment for antibiotic prescribing policy optimization under antimicrobial resistance
This paper addresses the critical challenge of quantitatively evaluating antibiotic prescribing policies under realistic uncertainty and partial obser...
-
PesTwin: a biology-informed Digital Twin for enabling precision farming
This paper addresses the critical bottleneck in precision agriculture: the inability to accurately forecast pest outbreaks in real-time, leading to su...
-
Equivariant Asynchronous Diffusion: An Adaptive Denoising Schedule for Accelerated Molecular Conformation Generation
This paper addresses the core challenge of generating physically plausible 3D molecular structures by bridging the gap between autoregressive methods ...
-
Omics Data Discovery Agents
This paper addresses the core challenge of making published omics data computationally reusable by automating the extraction, quantification, and inte...
-
Single-cell directional sensing at ultra-low chemoattractant concentrations from extreme first-passage events
This work addresses the core challenge of how a cell can rapidly and accurately determine the direction of a chemoattractant source when the signal is...
-
SDSR: A Spectral Divide-and-Conquer Approach for Species Tree Reconstruction
This paper addresses the computational bottleneck in reconstructing species trees from thousands of species and multiple genes by introducing a scalab...
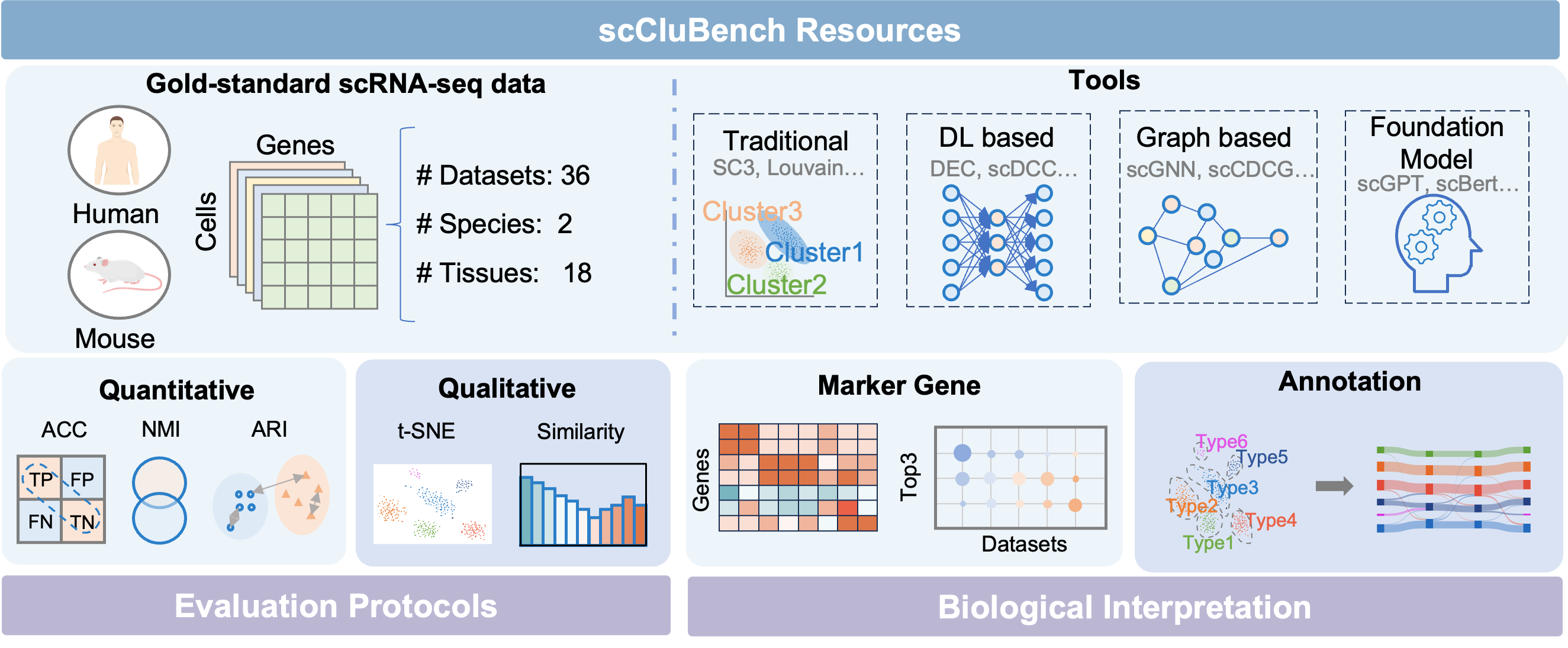
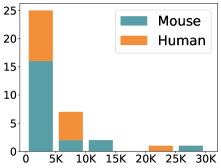
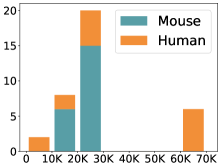
scCluBench: Comprehensive Benchmarking of Clustering Algorithms for Single-Cell RNA Sequencing
Not specified in provided content
30秒速读
IN SHORT: This paper addresses the critical gap of fragmented and non-standardized benchmarking in single-cell RNA-seq clustering, which hinders objective comparison and selection of appropriate methods for specific biological contexts.
核心创新
- Methodology Introduces scCluBench, the first comprehensive benchmarking framework that systematically evaluates 16 clustering methods across four categories (traditional, deep learning-based, graph-based, and foundation models) on 36 standardized datasets.
- Methodology Establishes standardized protocols for biological interpretation, including reproducible pipelines for marker gene identification and two distinct cell type annotation approaches (best-mapping and marker-overlap), validated with gold-standard references.
- Methodology Provides a unified and modular benchmarking workflow covering data preprocessing, clustering, and annotation with standardized input-output formats, ensuring reproducibility and fair comparison.
主要结论
- scCDCG (a cut-informed graph embedding model) achieved the highest average clustering accuracy (81.29 ± 1.45) across 36 datasets, outperforming other graph-based, deep learning, and traditional methods.
- Biological foundation models (scGPT, GeneFormer, GeneCompass) showed strong performance in classification tasks (e.g., scGPT achieved 98.14% ACC on Sapiens Ear Crista Ampullaris) but underperformed in direct clustering, highlighting a trade-off between general representation and task-specific optimization.
- The benchmark reveals method-specific limitations: traditional methods struggle with sparse data, deep learning models may fail to capture cell relationships, and graph-based models can suffer from over-smoothing, while most methods decouple embedding learning from clustering optimization.
摘要: Cell clustering is crucial for uncovering cellular heterogeneity in single-cell RNA sequencing (scRNA-seq) data by identifying cell types and marker genes. Despite its importance, benchmarks for scRNA-seq clustering methods remain fragmented, often lacking standardized protocols and failing to incorporate recent advances in artificial intelligence. To fill these gaps, we present scCluBench, a comprehensive benchmark of clustering algorithms for scRNA-seq data. First, scCluBench provides 36 scRNA-seq datasets collected from diverse public sources, covering multiple tissues, which are uniformly processed and standardized to ensure consistency for systematic evaluation and downstream analyses. To evaluate performance, we collect and reproduce a range of scRNA-seq clustering methods, including traditional, deep learning-based, graph-based, and biological foundation models. We comprehensively evaluate each method both quantitatively and qualitatively, using core performance metrics as well as visualization analyses. Furthermore, we construct representative downstream biological tasks, such as marker gene identification and cell type annotation, to further assess the practical utility. scCluBench then investigates the performance differences and applicability boundaries of various clustering models across diverse analytical tasks, systematically assessing their robustness and scalability in real-world scenarios. Overall, scCluBench offers a standardized and user-friendly benchmark for scRNA-seq clustering, with curated datasets, unified evaluation protocols, and transparent analyses, facilitating informed method selection and providing valuable insights into model generalizability and application scope.222All datasets, code, and the Extended version for scCluBench are available at the link: https://github.com/XPgogogo/scCluBench. More details for each stage are provided in the extended version.