
Paper List
-
SpikGPT: A High-Accuracy and Interpretable Spiking Attention Framework for Single-Cell Annotation
This paper addresses the core challenge of robust single-cell annotation across heterogeneous datasets with batch effects and the critical need to ide...
-
Unlocking hidden biomolecular conformational landscapes in diffusion models at inference time
This paper addresses the core challenge of efficiently and accurately sampling the conformational landscape of biomolecules from diffusion-based struc...
-
Personalized optimization of pediatric HD-tDCS for dose consistency and target engagement
This paper addresses the critical limitation of one-size-fits-all HD-tDCS protocols in pediatric populations by developing a personalized optimization...
-
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein con...
-
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
This paper addresses the core pain point of low sequence-structure alignment in existing synthetic datasets (e.g., AFDB), which severely limits the pe...
-
MoRSAIK: Sequence Motif Reactor Simulation, Analysis and Inference Kit in Python
This work addresses the computational bottleneck in simulating prebiotic RNA reactor dynamics by developing a Python package that tracks sequence moti...
-
On the Approximation of Phylogenetic Distance Functions by Artificial Neural Networks
This paper addresses the core challenge of developing computationally efficient and scalable neural network architectures that can learn accurate phyl...
-
EcoCast: A Spatio-Temporal Model for Continual Biodiversity and Climate Risk Forecasting
This paper addresses the critical bottleneck in conservation: the lack of timely, high-resolution, near-term forecasts of species distribution shifts ...
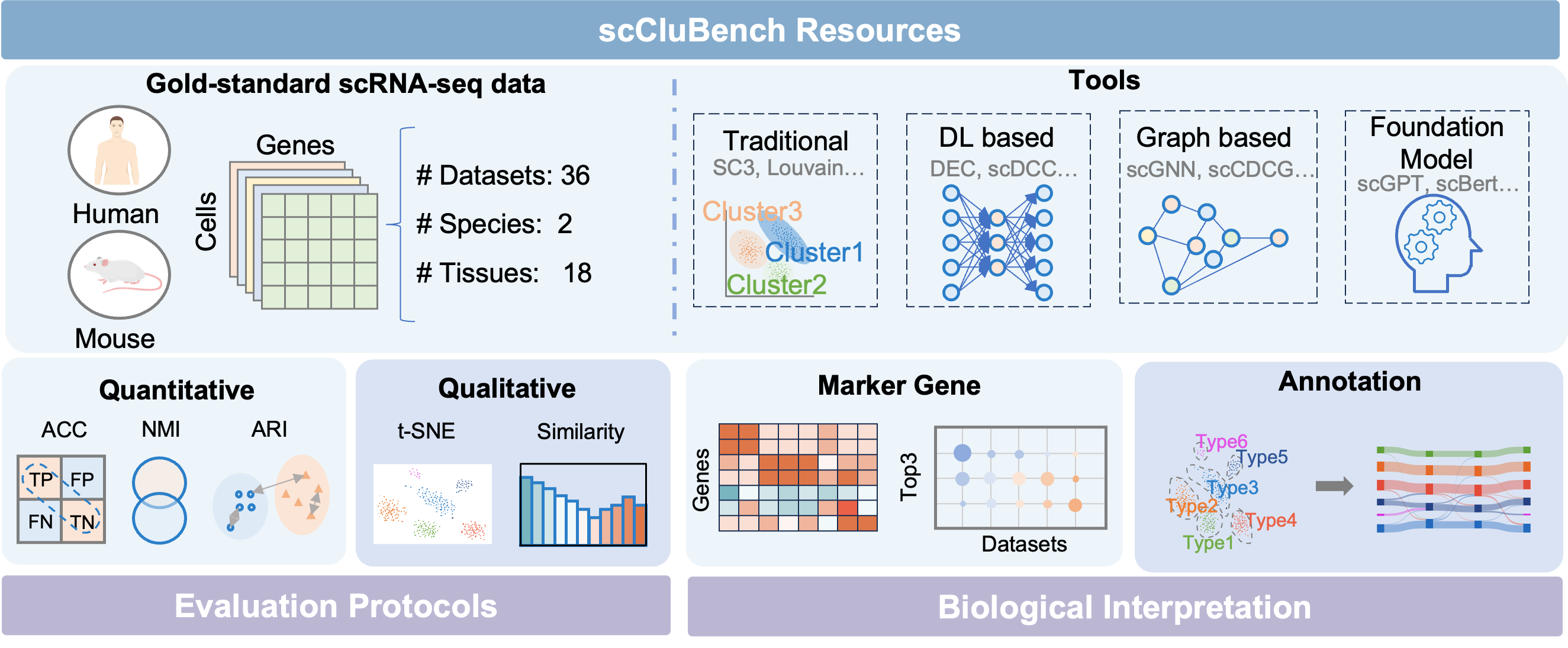
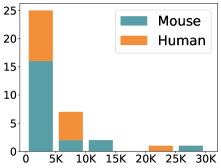
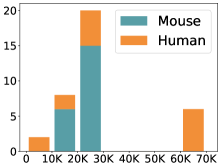
scCluBench: Comprehensive Benchmarking of Clustering Algorithms for Single-Cell RNA Sequencing
Not specified in provided content
30秒速读
IN SHORT: This paper addresses the critical gap of fragmented and non-standardized benchmarking in single-cell RNA-seq clustering, which hinders objective comparison and selection of appropriate methods for specific biological contexts.
核心创新
- Methodology Introduces scCluBench, the first comprehensive benchmarking framework that systematically evaluates 16 clustering methods across four categories (traditional, deep learning-based, graph-based, and foundation models) on 36 standardized datasets.
- Methodology Establishes standardized protocols for biological interpretation, including reproducible pipelines for marker gene identification and two distinct cell type annotation approaches (best-mapping and marker-overlap), validated with gold-standard references.
- Methodology Provides a unified and modular benchmarking workflow covering data preprocessing, clustering, and annotation with standardized input-output formats, ensuring reproducibility and fair comparison.
主要结论
- scCDCG (a cut-informed graph embedding model) achieved the highest average clustering accuracy (81.29 ± 1.45) across 36 datasets, outperforming other graph-based, deep learning, and traditional methods.
- Biological foundation models (scGPT, GeneFormer, GeneCompass) showed strong performance in classification tasks (e.g., scGPT achieved 98.14% ACC on Sapiens Ear Crista Ampullaris) but underperformed in direct clustering, highlighting a trade-off between general representation and task-specific optimization.
- The benchmark reveals method-specific limitations: traditional methods struggle with sparse data, deep learning models may fail to capture cell relationships, and graph-based models can suffer from over-smoothing, while most methods decouple embedding learning from clustering optimization.
摘要: Cell clustering is crucial for uncovering cellular heterogeneity in single-cell RNA sequencing (scRNA-seq) data by identifying cell types and marker genes. Despite its importance, benchmarks for scRNA-seq clustering methods remain fragmented, often lacking standardized protocols and failing to incorporate recent advances in artificial intelligence. To fill these gaps, we present scCluBench, a comprehensive benchmark of clustering algorithms for scRNA-seq data. First, scCluBench provides 36 scRNA-seq datasets collected from diverse public sources, covering multiple tissues, which are uniformly processed and standardized to ensure consistency for systematic evaluation and downstream analyses. To evaluate performance, we collect and reproduce a range of scRNA-seq clustering methods, including traditional, deep learning-based, graph-based, and biological foundation models. We comprehensively evaluate each method both quantitatively and qualitatively, using core performance metrics as well as visualization analyses. Furthermore, we construct representative downstream biological tasks, such as marker gene identification and cell type annotation, to further assess the practical utility. scCluBench then investigates the performance differences and applicability boundaries of various clustering models across diverse analytical tasks, systematically assessing their robustness and scalability in real-world scenarios. Overall, scCluBench offers a standardized and user-friendly benchmark for scRNA-seq clustering, with curated datasets, unified evaluation protocols, and transparent analyses, facilitating informed method selection and providing valuable insights into model generalizability and application scope.222All datasets, code, and the Extended version for scCluBench are available at the link: https://github.com/XPgogogo/scCluBench. More details for each stage are provided in the extended version.