
Paper List
-
Ill-Conditioning in Dictionary-Based Dynamic-Equation Learning: A Systems Biology Case Study
This paper addresses the critical challenge of numerical ill-conditioning and multicollinearity in library-based sparse regression methods (e.g., SIND...
-
Hybrid eTFCE–GRF: Exact Cluster-Size Retrieval with Analytical pp-Values for Voxel-Based Morphometry
This paper addresses the computational bottleneck in voxel-based neuroimaging analysis by providing a method that delivers exact cluster-size retrieva...
-
abx_amr_simulator: A simulation environment for antibiotic prescribing policy optimization under antimicrobial resistance
This paper addresses the critical challenge of quantitatively evaluating antibiotic prescribing policies under realistic uncertainty and partial obser...
-
PesTwin: a biology-informed Digital Twin for enabling precision farming
This paper addresses the critical bottleneck in precision agriculture: the inability to accurately forecast pest outbreaks in real-time, leading to su...
-
Equivariant Asynchronous Diffusion: An Adaptive Denoising Schedule for Accelerated Molecular Conformation Generation
This paper addresses the core challenge of generating physically plausible 3D molecular structures by bridging the gap between autoregressive methods ...
-
Omics Data Discovery Agents
This paper addresses the core challenge of making published omics data computationally reusable by automating the extraction, quantification, and inte...
-
Single-cell directional sensing at ultra-low chemoattractant concentrations from extreme first-passage events
This work addresses the core challenge of how a cell can rapidly and accurately determine the direction of a chemoattractant source when the signal is...
-
SDSR: A Spectral Divide-and-Conquer Approach for Species Tree Reconstruction
This paper addresses the computational bottleneck in reconstructing species trees from thousands of species and multiple genes by introducing a scalab...
On the Approximation of Phylogenetic Distance Functions by Artificial Neural Networks
Indiana University, Bloomington, IN 47405, USA
30秒速读
IN SHORT: This paper addresses the core challenge of developing computationally efficient and scalable neural network architectures that can learn accurate phylogenetic distance functions from simulated data, bridging the gap between simple distance methods and complex model-based inference.
核心创新
- Methodology Introduces minimal, permutation-invariant neural architectures (Sequence networks S and Pair networks P) specifically designed to approximate phylogenetic distance functions, ensuring invariance to taxa ordering without costly data augmentation.
- Methodology Leverages theoretical results from metric embedding (Bourgain's theorem, Johnson-Lindenstrauss Lemma) to inform network design, explicitly linking embedding dimension to the number of taxa for efficient representation.
- Methodology Demonstrates how equivariant layers and attention mechanisms can be structured to handle both i.i.d. and spatially correlated sequence data (e.g., models with indels or rate variation), adapting to the complexity of the generative evolutionary model.
主要结论
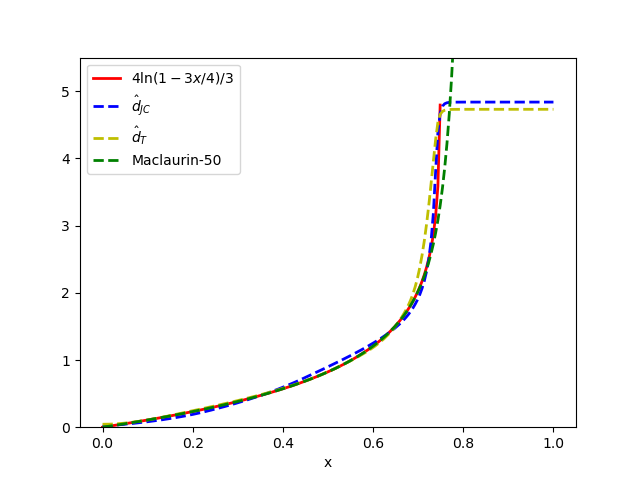
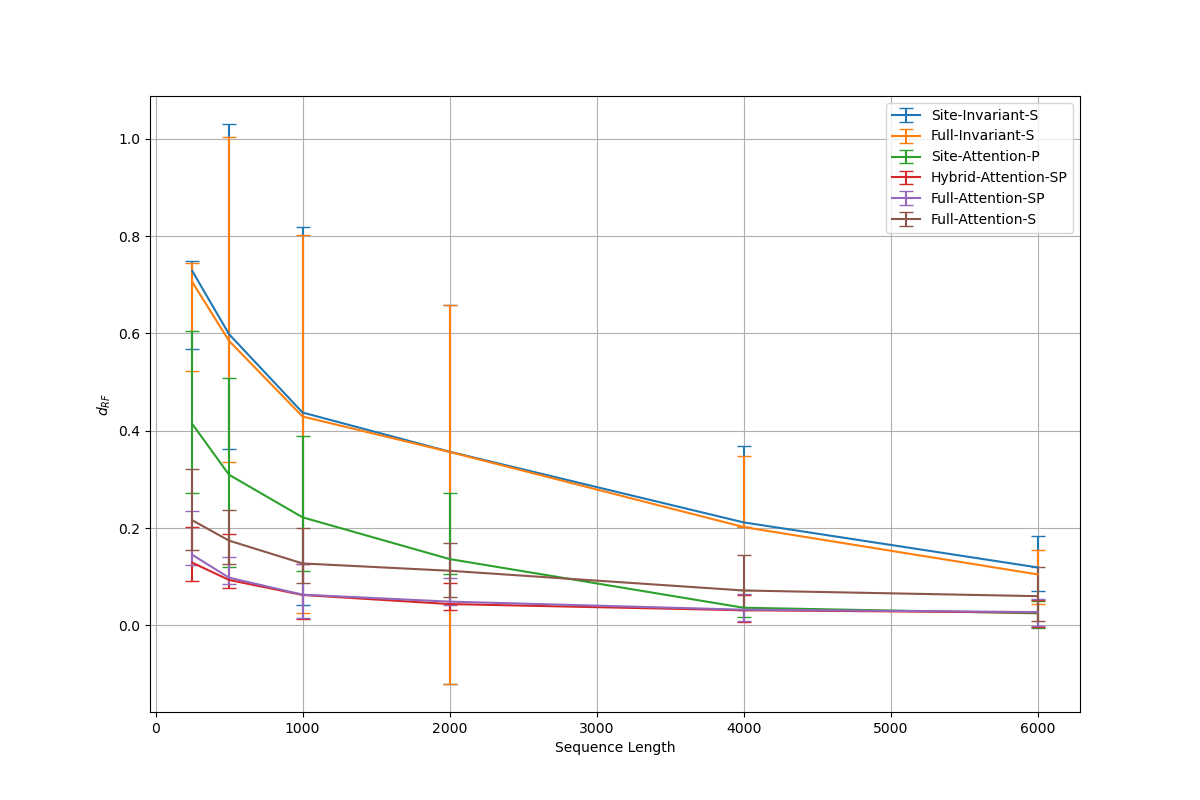
- The proposed minimal architectures (e.g., Sites-Invariant-S with ~7.6K parameters) achieve results comparable to state-of-the-art inference methods like IQ-TREE on simulated data under various models (JC, K2P, HKY, LG+indels), outperforming classic pairwise distance methods (d_H, d_JC, d_K2P) in most conditions.
- Architectures incorporating taxa-wise attention, while more memory-intensive, are necessary for complex evolutionary models with spatial dependencies; however, simpler networks suffice for simpler i.i.d. models, indicating an architecture-evolutionary model correspondence.
- Performance is highly sensitive to hyperparameters: validation error increases sharply with fewer than 4 attention heads or with hidden channel counts outside an optimal range (e.g., 32-128), aligning with theoretical requirements for learning graph-structured data.
摘要: Inferring the phylogenetic relationships among a sample of organisms is a fundamental problem in modern biology. While distance-based hierarchical clustering algorithms achieved early success on this task, these have been supplanted by Bayesian and maximum likelihood search procedures based on complex models of molecular evolution. In this work we describe minimal neural network architectures that can approximate classic phylogenetic distance functions and the properties required to learn distances under a variety of molecular evolutionary models. In contrast to model-based inference (and recently proposed model-free convolutional and transformer networks), these architectures have a small computational footprint and are scalable to large numbers of taxa and molecular characters. The learned distance functions generalize well and, given an appropriate training dataset, achieve results comparable to state-of-the art inference methods.