
Paper List
-
Nyxus: A Next Generation Image Feature Extraction Library for the Big Data and AI Era
This paper addresses the core pain point of efficiently extracting standardized, comparable features from massive (terabyte to petabyte-scale) biomedi...
-
Topological Enhancement of Protein Kinetic Stability
This work addresses the long-standing puzzle of why knotted proteins exist by demonstrating that deep knots provide a functional advantage through enh...
-
A Multi-Label Temporal Convolutional Framework for Transcription Factor Binding Characterization
This paper addresses the critical limitation of existing TF binding prediction methods that treat transcription factors as independent entities, faili...
-
Social Distancing Equilibria in Games under Conventional SI Dynamics
This paper solves the core problem of proving the existence and uniqueness of Nash equilibria in finite-duration SI epidemic games, showing they are a...
-
Binding Free Energies without Alchemy
This paper addresses the core bottleneck of computational expense in Absolute Binding Free Energy calculations by eliminating the need for numerous al...
-
SHREC: A Spectral Embedding-Based Approach for Ab-Initio Reconstruction of Helical Molecules
This paper addresses the core bottleneck in cryo-EM helical reconstruction: eliminating the dependency on accurate initial symmetry parameter estimati...
-
Budget-Sensitive Discovery Scoring: A Formally Verified Framework for Evaluating AI-Guided Scientific Selection
This paper addresses the critical gap in evaluating AI-guided scientific selection strategies under realistic budget constraints, where existing metri...
-
Probabilistic Joint and Individual Variation Explained (ProJIVE) for Data Integration
This paper addresses the core challenge of accurately decomposing shared (joint) and dataset-specific (individual) sources of variation in multi-modal...
On the Approximation of Phylogenetic Distance Functions by Artificial Neural Networks
Indiana University, Bloomington, IN 47405, USA
30秒速读
IN SHORT: This paper addresses the core challenge of developing computationally efficient and scalable neural network architectures that can learn accurate phylogenetic distance functions from simulated data, bridging the gap between simple distance methods and complex model-based inference.
核心创新
- Methodology Introduces minimal, permutation-invariant neural architectures (Sequence networks S and Pair networks P) specifically designed to approximate phylogenetic distance functions, ensuring invariance to taxa ordering without costly data augmentation.
- Methodology Leverages theoretical results from metric embedding (Bourgain's theorem, Johnson-Lindenstrauss Lemma) to inform network design, explicitly linking embedding dimension to the number of taxa for efficient representation.
- Methodology Demonstrates how equivariant layers and attention mechanisms can be structured to handle both i.i.d. and spatially correlated sequence data (e.g., models with indels or rate variation), adapting to the complexity of the generative evolutionary model.
主要结论
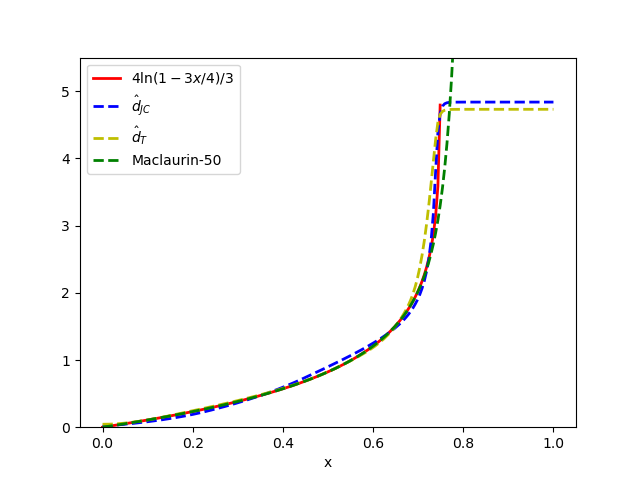
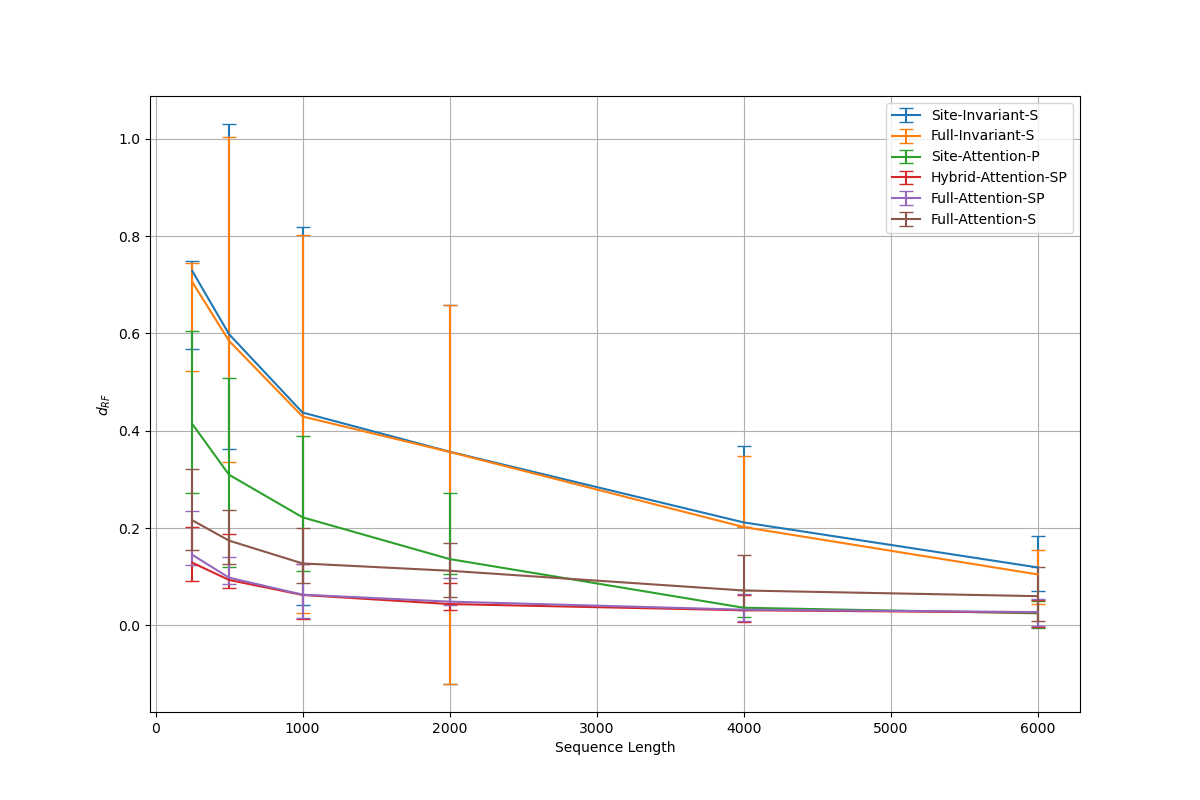
- The proposed minimal architectures (e.g., Sites-Invariant-S with ~7.6K parameters) achieve results comparable to state-of-the-art inference methods like IQ-TREE on simulated data under various models (JC, K2P, HKY, LG+indels), outperforming classic pairwise distance methods (d_H, d_JC, d_K2P) in most conditions.
- Architectures incorporating taxa-wise attention, while more memory-intensive, are necessary for complex evolutionary models with spatial dependencies; however, simpler networks suffice for simpler i.i.d. models, indicating an architecture-evolutionary model correspondence.
- Performance is highly sensitive to hyperparameters: validation error increases sharply with fewer than 4 attention heads or with hidden channel counts outside an optimal range (e.g., 32-128), aligning with theoretical requirements for learning graph-structured data.
摘要: Inferring the phylogenetic relationships among a sample of organisms is a fundamental problem in modern biology. While distance-based hierarchical clustering algorithms achieved early success on this task, these have been supplanted by Bayesian and maximum likelihood search procedures based on complex models of molecular evolution. In this work we describe minimal neural network architectures that can approximate classic phylogenetic distance functions and the properties required to learn distances under a variety of molecular evolutionary models. In contrast to model-based inference (and recently proposed model-free convolutional and transformer networks), these architectures have a small computational footprint and are scalable to large numbers of taxa and molecular characters. The learned distance functions generalize well and, given an appropriate training dataset, achieve results comparable to state-of-the art inference methods.