
Paper List
-
Evolutionarily Stable Stackelberg Equilibrium
通过要求追随者策略对突变入侵具有鲁棒性,弥合了斯塔克尔伯格领导力模型与演化稳定性之间的鸿沟。
-
Recovering Sparse Neural Connectivity from Partial Measurements: A Covariance-Based Approach with Granger-Causality Refinement
通过跨多个实验会话累积协方差统计,实现从部分记录到完整神经连接性的重建。
-
Atomic Trajectory Modeling with State Space Models for Biomolecular Dynamics
ATMOS通过提供一个基于SSM的高效框架,用于生物分子的原子级轨迹生成,弥合了计算昂贵的MD模拟与时间受限的深度生成模型之间的差距。
-
Slow evolution towards generalism in a model of variable dietary range
通过证明是种群统计噪声(而非确定性动力学)驱动了模式形成和泛化食性的演化,解决了间接竞争下物种形成的悖论。
-
Grounded Multimodal Retrieval-Augmented Drafting of Radiology Impressions Using Case-Based Similarity Search
通过将印象草稿基于检索到的历史病例,并采用明确引用和基于置信度的拒绝机制,解决放射学报告生成中的幻觉问题。
-
Unified Policy–Value Decomposition for Rapid Adaptation
通过双线性分解在策略和价值函数之间共享低维目标嵌入,实现对新颖任务的零样本适应。
-
Mathematical Modeling of Cancer–Bacterial Therapy: Analysis and Numerical Simulation via Physics-Informed Neural Networks
提供了一个严格的、无网格的PINN框架,用于模拟和分析细菌癌症疗法中复杂的、空间异质的相互作用。
-
Sample-Efficient Adaptation of Drug-Response Models to Patient Tumors under Strong Biological Domain Shift
通过从无标记分子谱中学习可迁移表征,利用最少的临床数据实现患者药物反应的有效预测。
On the Approximation of Phylogenetic Distance Functions by Artificial Neural Networks
Indiana University, Bloomington, IN 47405, USA
30秒速读
IN SHORT: This paper addresses the core challenge of developing computationally efficient and scalable neural network architectures that can learn accurate phylogenetic distance functions from simulated data, bridging the gap between simple distance methods and complex model-based inference.
核心创新
- Methodology Introduces minimal, permutation-invariant neural architectures (Sequence networks S and Pair networks P) specifically designed to approximate phylogenetic distance functions, ensuring invariance to taxa ordering without costly data augmentation.
- Methodology Leverages theoretical results from metric embedding (Bourgain's theorem, Johnson-Lindenstrauss Lemma) to inform network design, explicitly linking embedding dimension to the number of taxa for efficient representation.
- Methodology Demonstrates how equivariant layers and attention mechanisms can be structured to handle both i.i.d. and spatially correlated sequence data (e.g., models with indels or rate variation), adapting to the complexity of the generative evolutionary model.
主要结论
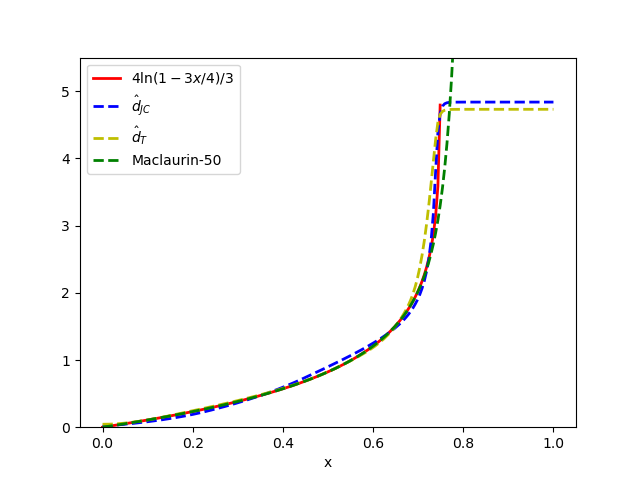
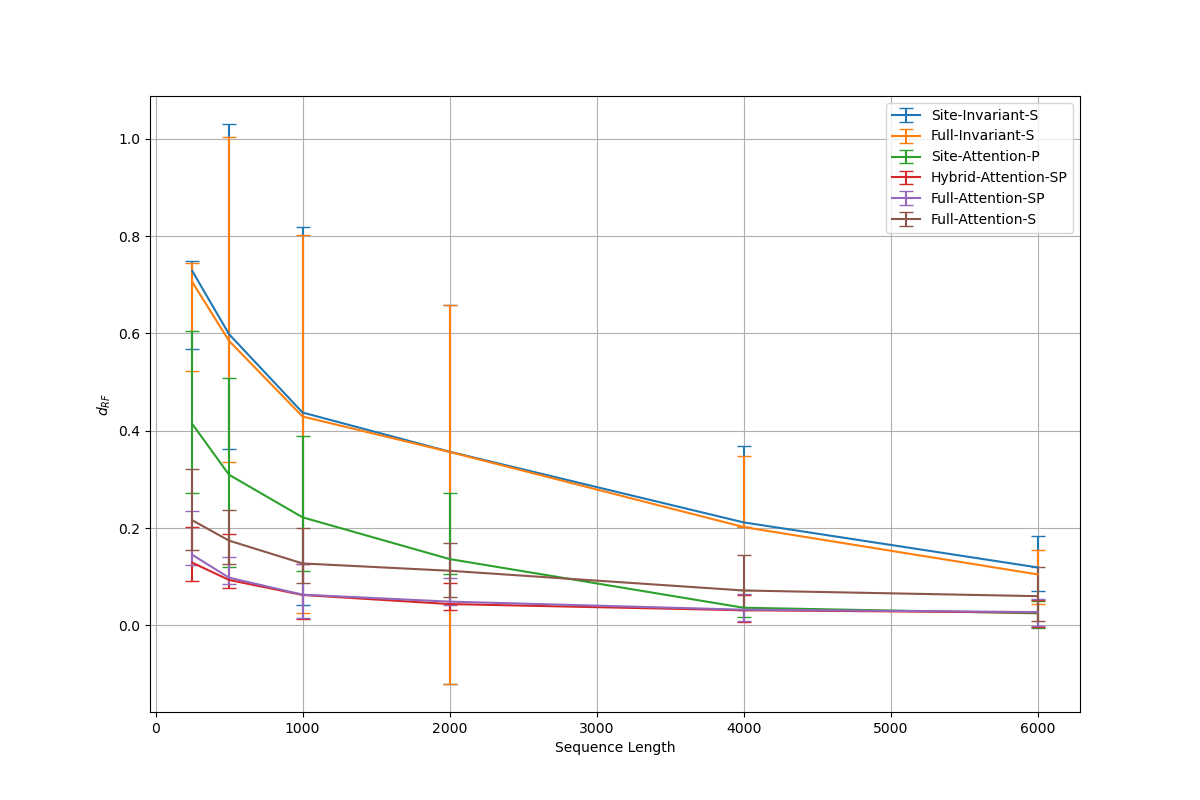
- The proposed minimal architectures (e.g., Sites-Invariant-S with ~7.6K parameters) achieve results comparable to state-of-the-art inference methods like IQ-TREE on simulated data under various models (JC, K2P, HKY, LG+indels), outperforming classic pairwise distance methods (d_H, d_JC, d_K2P) in most conditions.
- Architectures incorporating taxa-wise attention, while more memory-intensive, are necessary for complex evolutionary models with spatial dependencies; however, simpler networks suffice for simpler i.i.d. models, indicating an architecture-evolutionary model correspondence.
- Performance is highly sensitive to hyperparameters: validation error increases sharply with fewer than 4 attention heads or with hidden channel counts outside an optimal range (e.g., 32-128), aligning with theoretical requirements for learning graph-structured data.
摘要: Inferring the phylogenetic relationships among a sample of organisms is a fundamental problem in modern biology. While distance-based hierarchical clustering algorithms achieved early success on this task, these have been supplanted by Bayesian and maximum likelihood search procedures based on complex models of molecular evolution. In this work we describe minimal neural network architectures that can approximate classic phylogenetic distance functions and the properties required to learn distances under a variety of molecular evolutionary models. In contrast to model-based inference (and recently proposed model-free convolutional and transformer networks), these architectures have a small computational footprint and are scalable to large numbers of taxa and molecular characters. The learned distance functions generalize well and, given an appropriate training dataset, achieve results comparable to state-of-the art inference methods.