
Paper List
-
Evolutionarily Stable Stackelberg Equilibrium
通过要求追随者策略对突变入侵具有鲁棒性,弥合了斯塔克尔伯格领导力模型与演化稳定性之间的鸿沟。
-
Recovering Sparse Neural Connectivity from Partial Measurements: A Covariance-Based Approach with Granger-Causality Refinement
通过跨多个实验会话累积协方差统计,实现从部分记录到完整神经连接性的重建。
-
Atomic Trajectory Modeling with State Space Models for Biomolecular Dynamics
ATMOS通过提供一个基于SSM的高效框架,用于生物分子的原子级轨迹生成,弥合了计算昂贵的MD模拟与时间受限的深度生成模型之间的差距。
-
Slow evolution towards generalism in a model of variable dietary range
通过证明是种群统计噪声(而非确定性动力学)驱动了模式形成和泛化食性的演化,解决了间接竞争下物种形成的悖论。
-
Grounded Multimodal Retrieval-Augmented Drafting of Radiology Impressions Using Case-Based Similarity Search
通过将印象草稿基于检索到的历史病例,并采用明确引用和基于置信度的拒绝机制,解决放射学报告生成中的幻觉问题。
-
Unified Policy–Value Decomposition for Rapid Adaptation
通过双线性分解在策略和价值函数之间共享低维目标嵌入,实现对新颖任务的零样本适应。
-
Mathematical Modeling of Cancer–Bacterial Therapy: Analysis and Numerical Simulation via Physics-Informed Neural Networks
提供了一个严格的、无网格的PINN框架,用于模拟和分析细菌癌症疗法中复杂的、空间异质的相互作用。
-
Sample-Efficient Adaptation of Drug-Response Models to Patient Tumors under Strong Biological Domain Shift
通过从无标记分子谱中学习可迁移表征,利用最少的临床数据实现患者药物反应的有效预测。
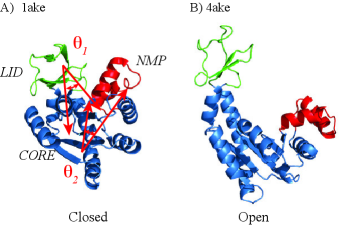
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
Department of Computer Science and Genome Center, University of California, Davis | Architecture et Dynamique des Macromolécules Biologiques, UMR 3528 du CNRS, Institut Pasteur | Department of Physics, School of Sciences, Great Bay University | Université Paris-Saclay, CNRS, CEA, Institut de Physique Théorique
30秒速读
IN SHORT: This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein conformations, a problem where existing methods often produce non-physical trajectories due to oversimplified energy surfaces and steric clashes.
核心创新
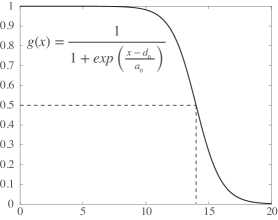
- Methodology Introduces SIDE (Stochastic Integro-Differential Equation), a novel Langevin bridge-based framework that efficiently approximates exact bridge equations at low temperatures to generate constrained transition trajectories.
- Methodology Develops a new coarse-grained potential that combines a Gō-like term (to preserve native backbone geometry) with a Rouse-type elastic energy term (from polymer physics), avoiding the problematic mixing of start/target conformation information used in prior methods like MinActionPath.
- Theory Provides a rigorous stochastic integro-differential formulation derived from the Langevin bridge formalism, which explicitly constrains trajectories to reach a target state within finite time, moving beyond Minimum Action Path (MAP) principles.
主要结论
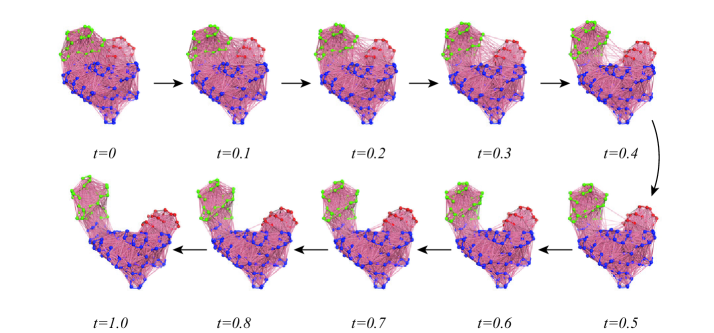
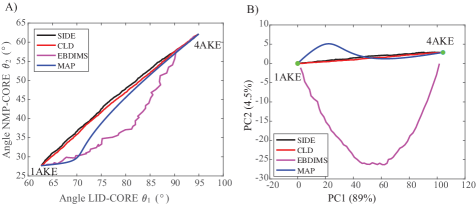
- The SIDE framework generates smooth, low-energy transition trajectories that maintain realistic molecular geometry, as demonstrated on several proteins undergoing large-scale conformational changes.
- SIDE frequently recovers experimentally supported intermediate states along transition paths, suggesting its paths have biological relevance beyond mere endpoint interpolation.
- Compared to established methods like MinActionPath and EBDIMS, SIDE offers improved physical realism and computational efficiency for modeling biomolecular conformational transitions, though challenges remain for highly complex motions.
摘要: We introduce a computational framework for generating realistic transition paths between distinct conformations of large biomolecular systems. The method is built on a stochastic integro-differential formulation derived from the Langevin bridge formalism, which constrains molecular trajectories to reach a prescribed final state within a finite time and yields an efficient low-temperature approximation of the exact bridge equation. To obtain physically meaningful protein transitions, we couple this formulation to a new coarse-grained potential combining a Gō-like term that preserves native backbone geometry with a Rouse-type elastic energy term from polymer physics; we refer to the resulting approach as SIDE. We evaluate SIDE on several proteins undergoing large-scale conformational changes and compare its performance with established methods such as MinActionPath and EBDIMS. SIDE generates smooth, low-energy trajectories that maintain molecular geometry and frequently recover experimentally supported intermediate states. Although challenges remain for highly complex motions—largely due to the simplified coarse-grained potential—our results demonstrate that SIDE offers a powerful and computationally efficient strategy for modeling biomolecular conformational transitions.