
Paper List
-
Nyxus: A Next Generation Image Feature Extraction Library for the Big Data and AI Era
This paper addresses the core pain point of efficiently extracting standardized, comparable features from massive (terabyte to petabyte-scale) biomedi...
-
Topological Enhancement of Protein Kinetic Stability
This work addresses the long-standing puzzle of why knotted proteins exist by demonstrating that deep knots provide a functional advantage through enh...
-
A Multi-Label Temporal Convolutional Framework for Transcription Factor Binding Characterization
This paper addresses the critical limitation of existing TF binding prediction methods that treat transcription factors as independent entities, faili...
-
Social Distancing Equilibria in Games under Conventional SI Dynamics
This paper solves the core problem of proving the existence and uniqueness of Nash equilibria in finite-duration SI epidemic games, showing they are a...
-
Binding Free Energies without Alchemy
This paper addresses the core bottleneck of computational expense in Absolute Binding Free Energy calculations by eliminating the need for numerous al...
-
SHREC: A Spectral Embedding-Based Approach for Ab-Initio Reconstruction of Helical Molecules
This paper addresses the core bottleneck in cryo-EM helical reconstruction: eliminating the dependency on accurate initial symmetry parameter estimati...
-
Budget-Sensitive Discovery Scoring: A Formally Verified Framework for Evaluating AI-Guided Scientific Selection
This paper addresses the critical gap in evaluating AI-guided scientific selection strategies under realistic budget constraints, where existing metri...
-
Probabilistic Joint and Individual Variation Explained (ProJIVE) for Data Integration
This paper addresses the core challenge of accurately decomposing shared (joint) and dataset-specific (individual) sources of variation in multi-modal...
Simulation and inference methods for non-Markovian stochastic biochemical reaction networks
School of Mathematical Sciences, Queensland University of Technology | Centre for Data Science, Queensland University of Technology | ARC Centre of Excellence for Mathematical Analysis of Cellular Systems (MACSYS), Queensland University of Technology
30秒速读
IN SHORT: This paper addresses the computational bottleneck of simulating and performing Bayesian inference for non-Markovian biochemical systems with history-dependent delays, which are crucial for modeling processes like gene transcription but are prohibitively expensive with existing methods.
核心创新
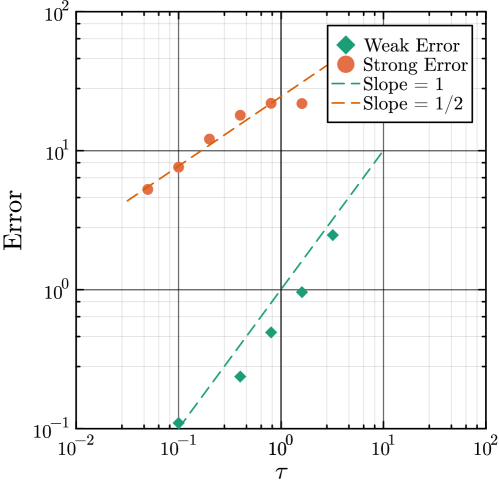
- Methodology Generalizes the next reaction method and τ-leaping to support arbitrary inter-event time distributions for non-Markovian systems, maintaining computational scalability.
- Methodology Introduces a novel coupling scheme to generate positively correlated exact and approximate non-Markovian sample paths, a prerequisite for variance reduction techniques.
- Methodology Enables the application of multifidelity and multilevel Monte Carlo (MLMC) methods to non-Markovian systems for the first time, bridging a significant methodological gap.
主要结论
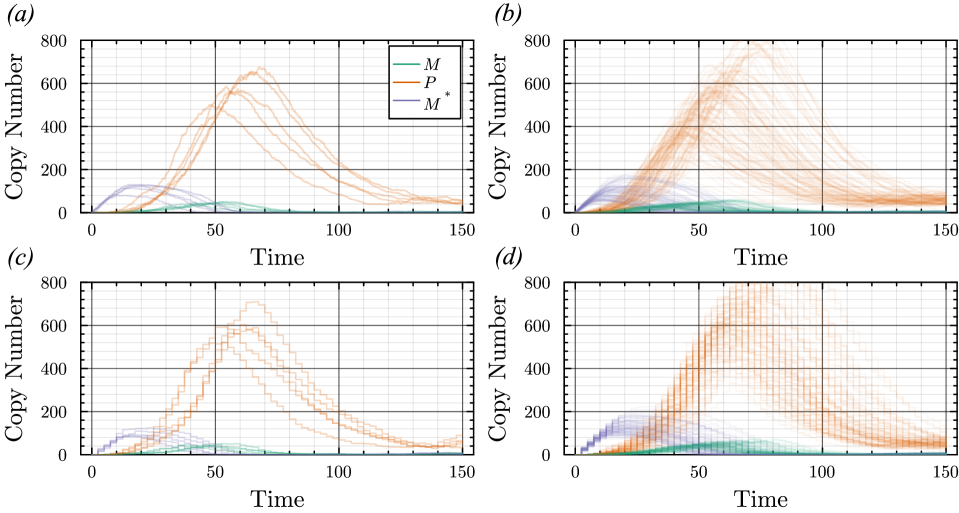
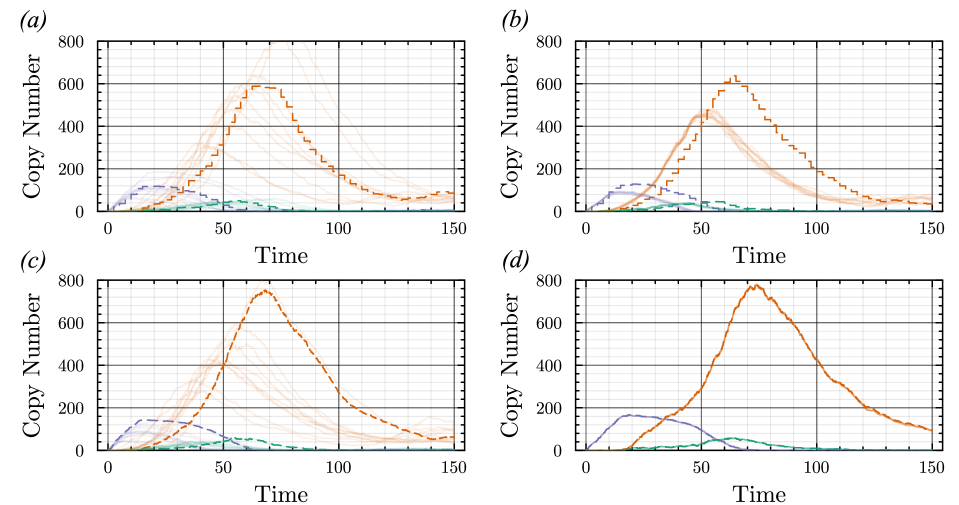
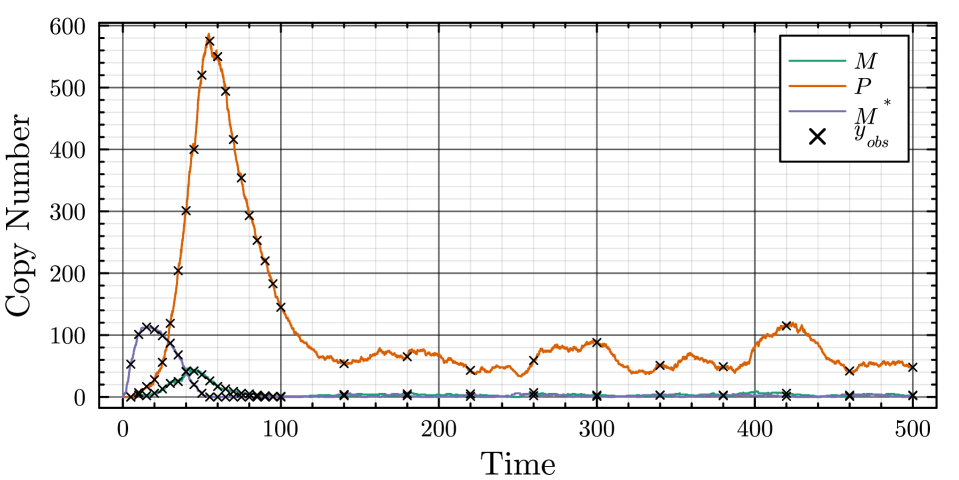
- The proposed non-Markovian simulation algorithms and coupling scheme successfully enable multifidelity inference, demonstrated on a gene regulation model with delayed auto-inhibition.
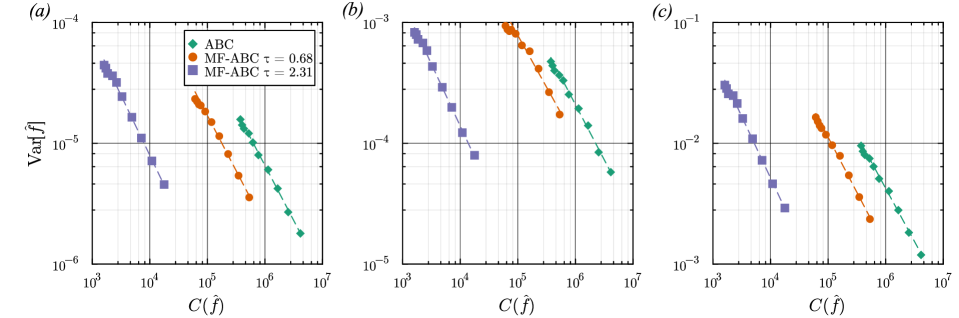
- The method achieves a computational speedup of two orders of magnitude (100x) in inference efficiency compared to standard approaches for the non-Markovian case study.
- The framework supports arbitrary delay distributions (state- and time-dependent), significantly extending the practical modeling scope beyond previous methods limited to simpler, time-only delays.
摘要: Stochastic models of biochemical reaction networks are widely used to capture intrinsic noise in cellular systems. The typical formulation of these models are based on Markov processes for which there is extensive research on efficient simulation and inference. However, there are biological processes, such as gene transcription and translation, that introduce history dependent dynamics requiring non-Markovian processes to accurately capture the stochastic dynamics of the system. This greater realism comes with additional computational challenges for simulation and parameter inference. We develop efficient stochastic simulation algorithms for well-mixed non-Markovian stochastic biochemical reaction networks with delays that depend on system state and time. Our methods generalize the next reaction method and τ-leaping method to support arbitrary inter-event time distributions while preserving computational scalability. We also introduce a coupling scheme to generate exact non-Markovian sample paths that are positively correlated to an approximate non-Markovian τ-leaping sample path. This enables substantial computational gains for Bayesian inference of model parameters though multifidelity simulation-based inference schemes. We demonstrate the effectiveness of our approach on a gene regulation model with delayed auto-inhibition, showing substantial gains in both simulation accuracy and inference efficiency of two orders of magnitude. These results extend the practical applicability of non-Markovian models in systems biology and beyond.