
Paper List
-
Discovery of a Hematopoietic Manifold in scGPT Yields a Method for Extracting Performant Algorithms from Biological Foundation Model Internals
This work addresses the core challenge of extracting reusable, interpretable, and high-performance biological algorithms from the opaque internal repr...
-
MS2MetGAN: Latent-space adversarial training for metabolite–spectrum matching in MS/MS database search
This paper addresses the critical bottleneck in metabolite identification: the generation of high-quality negative training samples that are structura...
-
Toward Robust, Reproducible, and Widely Accessible Intracranial Language Brain-Computer Interfaces: A Comprehensive Review of Neural Mechanisms, Hardware, Algorithms, Evaluation, Clinical Pathways and Future Directions
This review addresses the core challenge of fragmented and heterogeneous evidence that hinders the clinical translation of intracranial language BCIs,...
-
Less Is More in Chemotherapy of Breast Cancer
通过纳入细胞周期时滞和竞争项,解决了现有肿瘤-免疫模型的过度简化问题,以定量比较化疗方案。
-
Fold-CP: A Context Parallelism Framework for Biomolecular Modeling
This paper addresses the critical bottleneck of GPU memory limitations that restrict AlphaFold 3-like models to processing only a few thousand residue...
-
Open Biomedical Knowledge Graphs at Scale: Construction, Federation, and AI Agent Access with Samyama Graph Database
This paper addresses the core pain point of fragmented biomedical data by constructing and federating large-scale, open knowledge graphs to enable sea...
-
Predictive Analytics for Foot Ulcers Using Time-Series Temperature and Pressure Data
This paper addresses the critical need for continuous, real-time monitoring of diabetic foot health by developing an unsupervised anomaly detection fr...
-
Hypothesis-Based Particle Detection for Accurate Nanoparticle Counting and Digital Diagnostics
This paper addresses the core challenge of achieving accurate, interpretable, and training-free nanoparticle counting in digital diagnostic assays, wh...
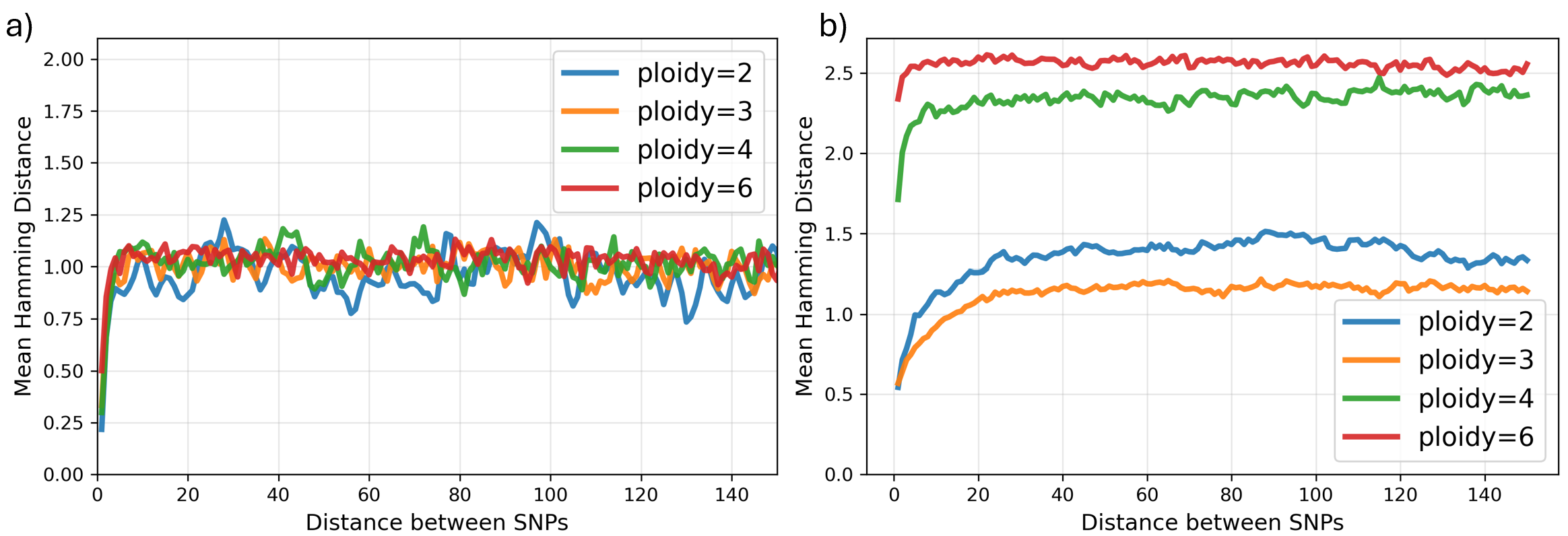
pHapCompass: Probabilistic Assembly and Uncertainty Quantification of Polyploid Haplotype Phase
School of Computing, University of Connecticut | Department of Entomology and Plant Pathology, University of Tennessee | Institute for Systems Genomics, University of Connecticut
30秒速读
IN SHORT: This paper addresses the core challenge of accurately assembling polyploid haplotypes from sequencing data, where read assignment ambiguity and an exponential search space of possible phasings have hindered reliable reconstruction and uncertainty quantification.
核心创新
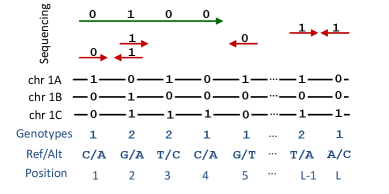
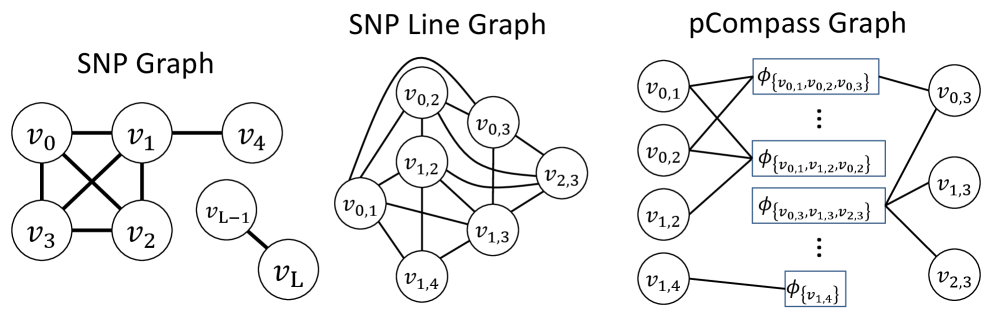
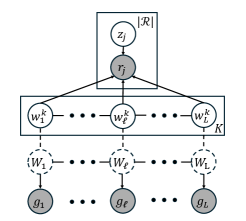
- Methodology Introduces pHapCompass, the first probabilistic haplotype assembler for diploid and polyploid genomes that explicitly models read assignment ambiguity to compute a distribution over haplotype phasings, enabling formal uncertainty quantification.
- Methodology Develops two distinct graph-theoretic algorithms: pHapCompass-short (a Markov random field for high-coverage short reads) and pHapCompass-long (a hierarchical mixture model for low-coverage long reads), both designed to scale with genomic complexity.
- Methodology Creates the first computational workflow for simulating realistic auto- and allopolyploid genomes and sequencing data, addressing a critical gap in benchmarking tools that previously relied on oversimplified synthetic genomes.
主要结论
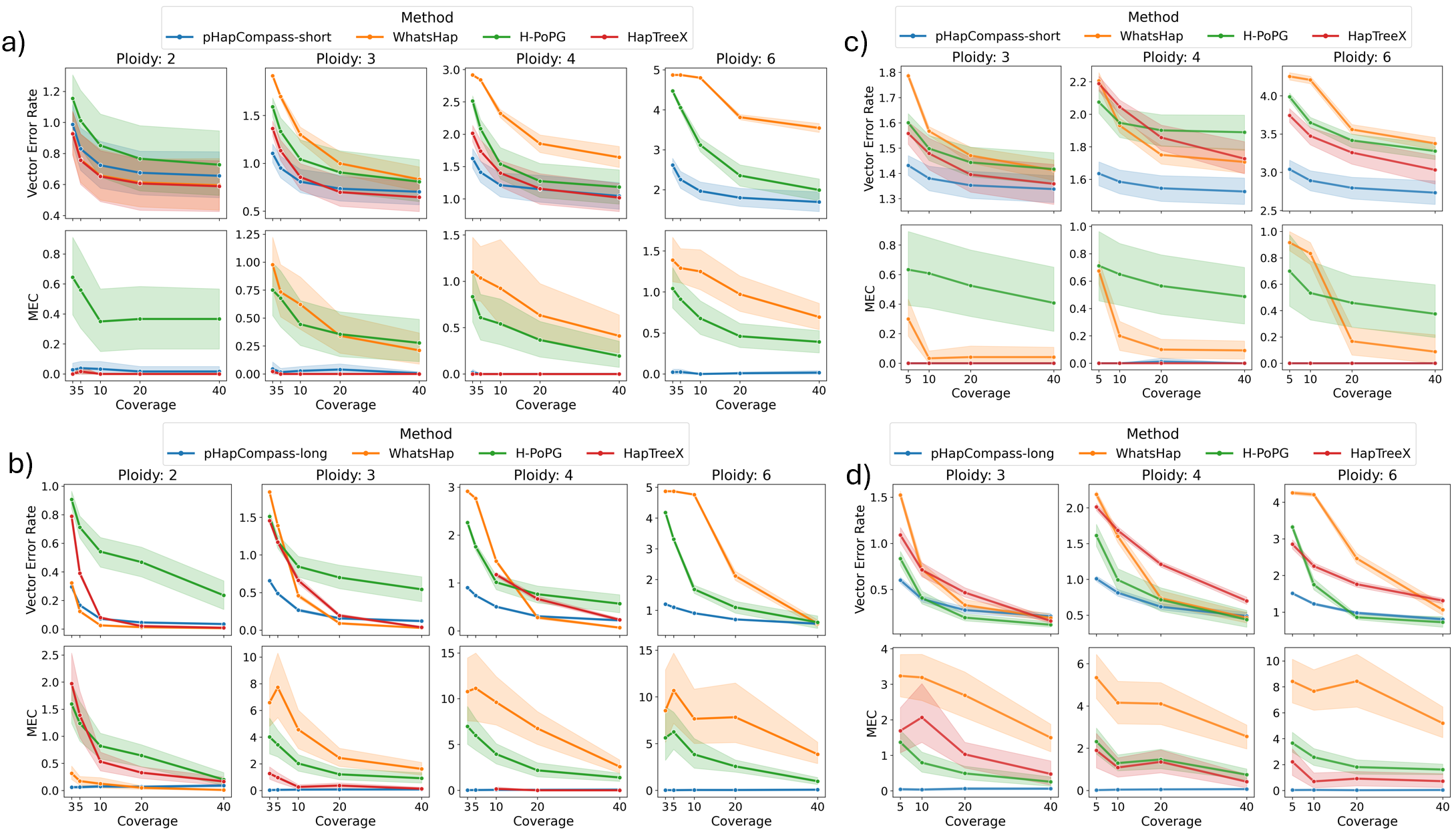
- pHapCompass demonstrates competitive performance against existing assemblers across varying ploidy levels, coverage depths, and mutation rates, while uniquely providing accurate quantification of phase uncertainty.
- The developed simulation workflow generates more realistic benchmarking datasets, revealing that prior methods often overestimate performance on simplistic synthetic genomes.
- The framework successfully assembled an allo-octoploid strawberry chromosome, showcasing practical applicability to complex, real-world polyploid genomes.
摘要: Computing haplotypes from sequencing data, i.e. haplotype assembly, is an important component of foundational molecular and population genetics problems, including interpreting the effects of genetic variation on complex traits and reconstructing genealogical relationships. Assembling the haplotypes of polyploid genomes remains a significant challenge due to the exponential search space of haplotype phasings and read assignment ambiguity; the latter challenge is particularly difficult for polyploid haplotype assemblers since the information contained within the observed sequence reads is often insufficient for unambiguous haplotype assignment in polyploid genomes. We present pHapCompass, probabilistic haplotype assembly algorithms for diploid and polyploid genomes that explicitly model and propagate read assignment ambiguity to compute a distribution over polyploid haplotype phasings. We develop graph theoretic algorithms to enable statistical inference and uncertainty quantification despite an exponential space of possible phasings. Since prior work evaluates polyploid haplotype assembly on synthetic genomes that do not reflect the realistic genomic complexity of polyploidy organisms, we develop a computational workflow for simulating genomes and DNA-seq for auto- and allopolyploids. Additionally, we generalize the vector error rate and minimum error correction evaluation criteria for partially phased haplotypes. Benchmarking of pHapCompass and several existing polyploid haplotype assemblers shows that pHapCompass yields competitive performance across varying genomic complexities and polyploid structures while retaining an accurate quantification of phase uncertainty. The source code for pHapCompass, simulation scripts, and datasets are freely available at https://github.com/bayesomicslab/pHapCompass.