
Paper List
-
Formation of Artificial Neural Assemblies by Biologically Plausible Inhibition Mechanisms
This work addresses the core limitation of the Assembly Calculus model—its fixed-size, biologically implausible k-WTA selection process—by introducing...
-
How to make the most of your masked language model for protein engineering
This paper addresses the critical bottleneck of efficiently sampling high-quality, diverse protein sequences from Masked Language Models (MLMs) for pr...
-
Module control in youth symptom networks across COVID-19
This paper addresses the core challenge of distinguishing whether a prolonged societal stressor (COVID-19) fundamentally reorganizes the architecture ...
-
JEDI: Jointly Embedded Inference of Neural Dynamics
This paper addresses the core challenge of inferring context-dependent neural dynamics from noisy, high-dimensional recordings using a single unified ...
-
ATP Level and Phosphorylation Free Energy Regulate Trigger-Wave Speed and Critical Nucleus Size in Cellular Biochemical Systems
This work addresses the core challenge of quantitatively predicting how the cellular energy state (ATP level and phosphorylation free energy) governs ...
-
Packaging Jupyter notebooks as installable desktop apps using LabConstrictor
This paper addresses the core pain point of ensuring Jupyter notebook reproducibility and accessibility across different computing environments, parti...
-
SNPgen: Phenotype-Supervised Genotype Representation and Synthetic Data Generation via Latent Diffusion
This paper addresses the core challenge of generating privacy-preserving synthetic genotype data that maintains both statistical fidelity and downstre...
-
Continuous Diffusion Transformers for Designing Synthetic Regulatory Elements
This paper addresses the challenge of efficiently generating novel, cell-type-specific regulatory DNA sequences with high predicted activity while min...
Unlocking hidden biomolecular conformational landscapes in diffusion models at inference time
Stanford University | Yale School of Medicine

30秒速读
IN SHORT: This paper addresses the core challenge of efficiently and accurately sampling the conformational landscape of biomolecules from diffusion-based structure prediction models, which typically output highly concentrated distributions around a single static structure.
核心创新
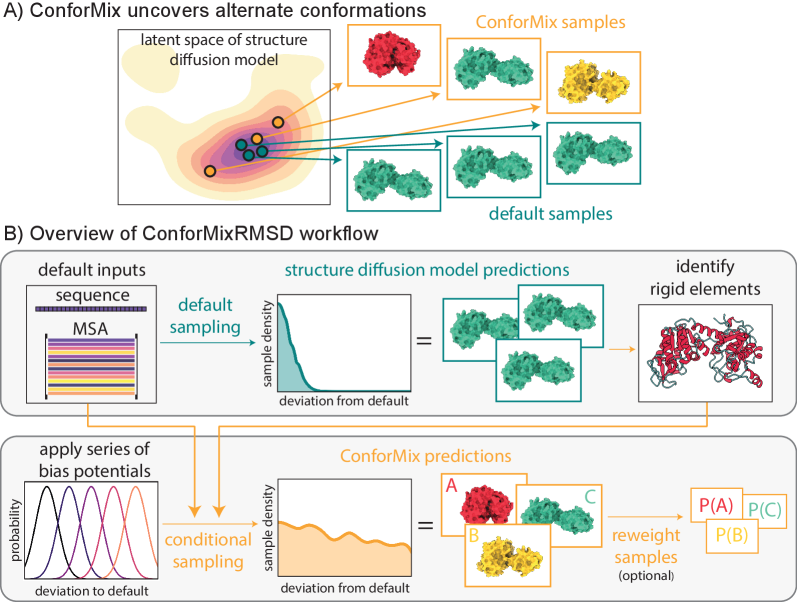
- Methodology Introduces ConforMix, a novel inference-time algorithm combining twisted sequential Monte Carlo (SMC) with automated exploration of the diffusion landscape, enabling asymptotically exact sampling of conditional distributions without additional model training.
- Methodology Presents ConforMixRMSD, an instantiation for automated exploration that biases sampling away from the default prediction using RMSD-based potentials on rigid secondary structure elements, recovering diverse conformations without prior knowledge of degrees of freedom.
- Methodology Applies the multistate Bennett acceptance ratio (MBAR) free energy estimation algorithm to diffusion models for the first time, enabling reconstruction of the unbiased model landscape from conditional samples.
主要结论
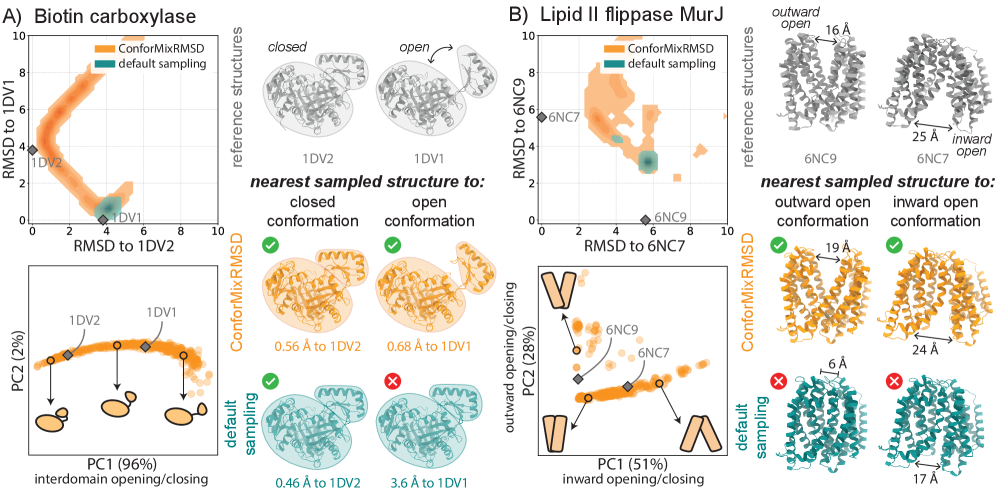
- ConforMixRMSD applied to Boltz-1 (an AlphaFold 3-like model) significantly outperforms MSA-modification baselines (AFCluster, AFSample2, CF-random) in recovering experimentally observed alternative conformations for domain motion (coverage: 0.69 ± 0.15 vs. 0.51 ± 0.17 for best baseline), membrane transporter (0.33 ± 0.23 vs. 0.20 ± 0.20), and cryptic pocket (0.45 ± 0.18 vs. 0.39 ± 0.16) protein sets, as measured by coverage at 50% of reference-to-reference RMSD.
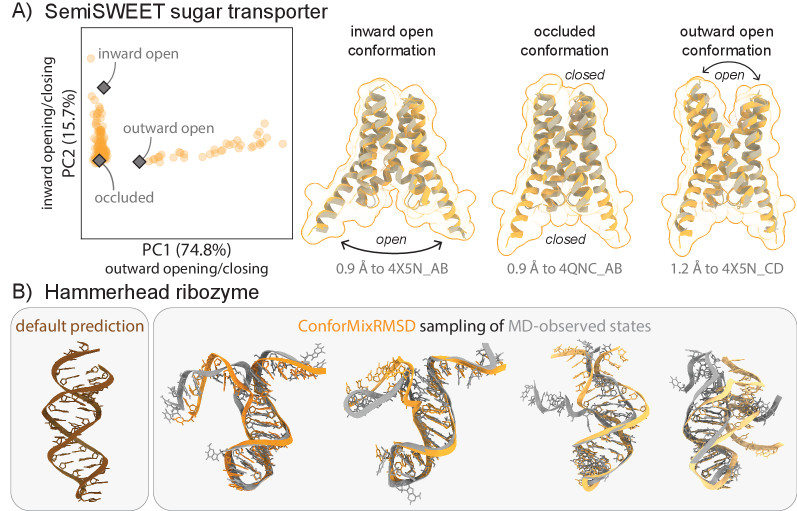
- The method captures biologically relevant conformational transitions (domain motion, transporter cycling, cryptic pocket flexibility) while avoiding unphysical states through filtering based on pLDDT values and clash detection, demonstrating its utility for exploring continuous transitions.
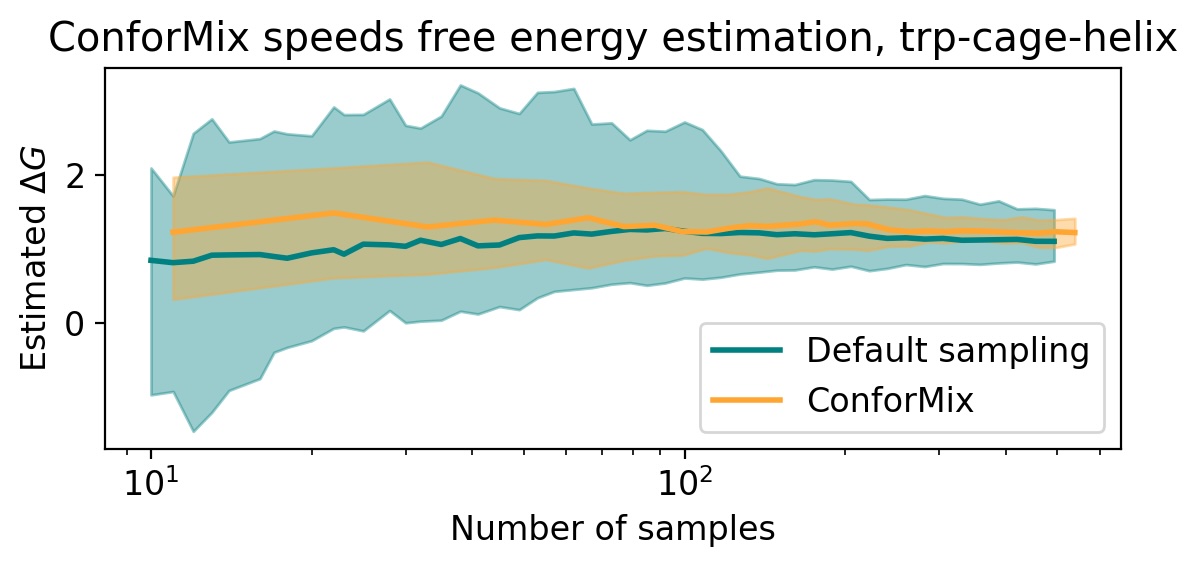
- ConforMix enables efficient free energy estimation when applied to models like BioEmu, boosting the speed of such calculations, and its framework is orthogonal to model pretraining improvements, meaning it would benefit even a hypothetical model that perfectly reproduces the Boltzmann distribution.
摘要: The function of biomolecules such as proteins depends on their ability to interconvert between a wide range of structures or “conformations.” Researchers have endeavored for decades to develop computational methods to predict the distribution of conformations, which is far harder to determine experimentally than a static folded structure. We present ConforMix, an inference-time algorithm that enhances sampling of conformational distributions using a combination of classifier guidance, filtering, and free energy estimation. Our approach upgrades diffusion models—whether trained for static structure prediction or conformational generation—to enable more efficient discovery of conformational variability without requiring prior knowledge of major degrees of freedom. ConforMix is orthogonal to improvements in model pretraining and would benefit even a hypothetical model that perfectly reproduced the Boltzmann distribution. Remarkably, when applied to a diffusion model trained for static structure prediction, ConforMix captures structural changes including domain motion, cryptic pocket flexibility, and transporter cycling, while avoiding unphysical states. Case studies of biologically critical proteins demonstrate the scalability, accuracy, and utility of this method.