
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
Unlocking hidden biomolecular conformational landscapes in diffusion models at inference time
Stanford University | Yale School of Medicine

30秒速读
IN SHORT: This paper addresses the core challenge of efficiently and accurately sampling the conformational landscape of biomolecules from diffusion-based structure prediction models, which typically output highly concentrated distributions around a single static structure.
核心创新
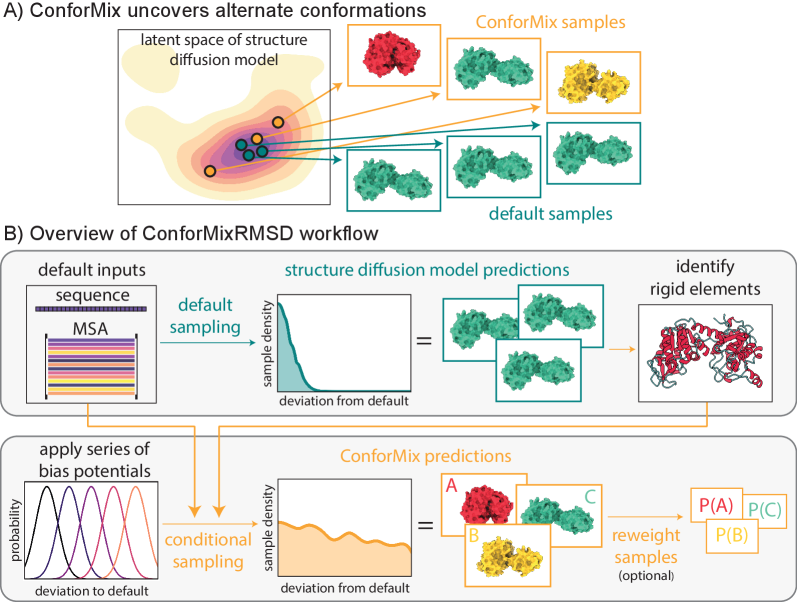
- Methodology Introduces ConforMix, a novel inference-time algorithm combining twisted sequential Monte Carlo (SMC) with automated exploration of the diffusion landscape, enabling asymptotically exact sampling of conditional distributions without additional model training.
- Methodology Presents ConforMixRMSD, an instantiation for automated exploration that biases sampling away from the default prediction using RMSD-based potentials on rigid secondary structure elements, recovering diverse conformations without prior knowledge of degrees of freedom.
- Methodology Applies the multistate Bennett acceptance ratio (MBAR) free energy estimation algorithm to diffusion models for the first time, enabling reconstruction of the unbiased model landscape from conditional samples.
主要结论
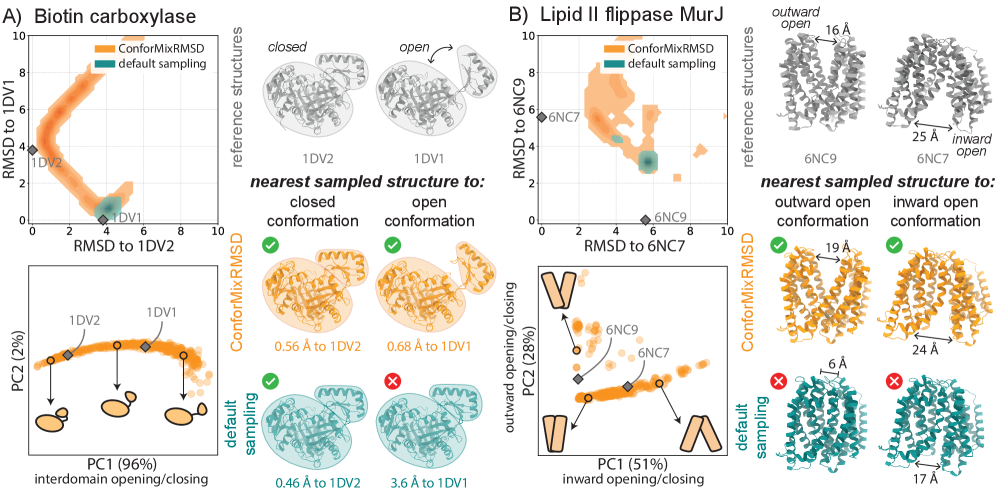
- ConforMixRMSD applied to Boltz-1 (an AlphaFold 3-like model) significantly outperforms MSA-modification baselines (AFCluster, AFSample2, CF-random) in recovering experimentally observed alternative conformations for domain motion (coverage: 0.69 ± 0.15 vs. 0.51 ± 0.17 for best baseline), membrane transporter (0.33 ± 0.23 vs. 0.20 ± 0.20), and cryptic pocket (0.45 ± 0.18 vs. 0.39 ± 0.16) protein sets, as measured by coverage at 50% of reference-to-reference RMSD.
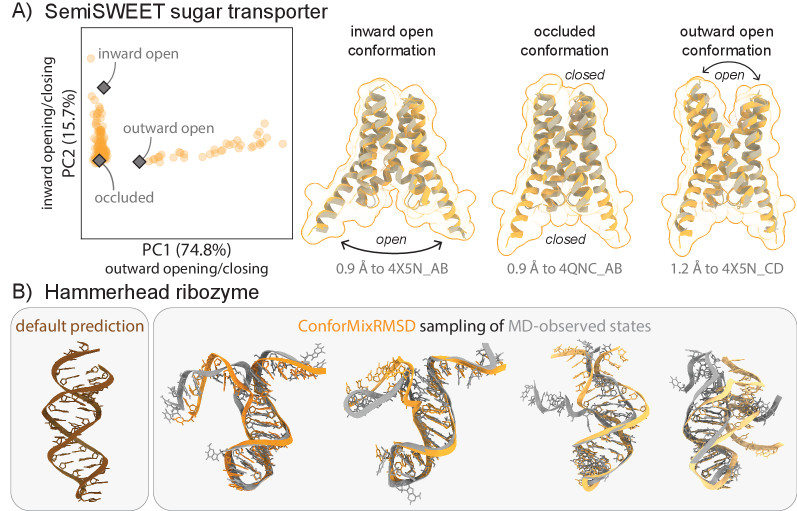
- The method captures biologically relevant conformational transitions (domain motion, transporter cycling, cryptic pocket flexibility) while avoiding unphysical states through filtering based on pLDDT values and clash detection, demonstrating its utility for exploring continuous transitions.
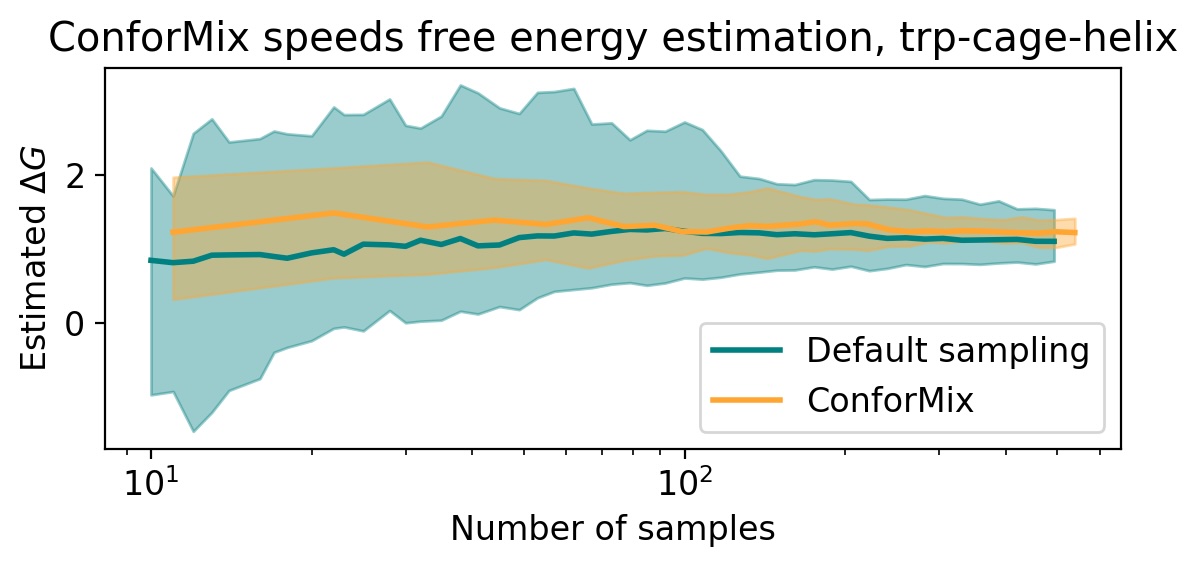
- ConforMix enables efficient free energy estimation when applied to models like BioEmu, boosting the speed of such calculations, and its framework is orthogonal to model pretraining improvements, meaning it would benefit even a hypothetical model that perfectly reproduces the Boltzmann distribution.
摘要: The function of biomolecules such as proteins depends on their ability to interconvert between a wide range of structures or “conformations.” Researchers have endeavored for decades to develop computational methods to predict the distribution of conformations, which is far harder to determine experimentally than a static folded structure. We present ConforMix, an inference-time algorithm that enhances sampling of conformational distributions using a combination of classifier guidance, filtering, and free energy estimation. Our approach upgrades diffusion models—whether trained for static structure prediction or conformational generation—to enable more efficient discovery of conformational variability without requiring prior knowledge of major degrees of freedom. ConforMix is orthogonal to improvements in model pretraining and would benefit even a hypothetical model that perfectly reproduced the Boltzmann distribution. Remarkably, when applied to a diffusion model trained for static structure prediction, ConforMix captures structural changes including domain motion, cryptic pocket flexibility, and transporter cycling, while avoiding unphysical states. Case studies of biologically critical proteins demonstrate the scalability, accuracy, and utility of this method.