
Paper List
-
Discovery of a Hematopoietic Manifold in scGPT Yields a Method for Extracting Performant Algorithms from Biological Foundation Model Internals
This work addresses the core challenge of extracting reusable, interpretable, and high-performance biological algorithms from the opaque internal repr...
-
MS2MetGAN: Latent-space adversarial training for metabolite–spectrum matching in MS/MS database search
This paper addresses the critical bottleneck in metabolite identification: the generation of high-quality negative training samples that are structura...
-
Toward Robust, Reproducible, and Widely Accessible Intracranial Language Brain-Computer Interfaces: A Comprehensive Review of Neural Mechanisms, Hardware, Algorithms, Evaluation, Clinical Pathways and Future Directions
This review addresses the core challenge of fragmented and heterogeneous evidence that hinders the clinical translation of intracranial language BCIs,...
-
Less Is More in Chemotherapy of Breast Cancer
通过纳入细胞周期时滞和竞争项,解决了现有肿瘤-免疫模型的过度简化问题,以定量比较化疗方案。
-
Fold-CP: A Context Parallelism Framework for Biomolecular Modeling
This paper addresses the critical bottleneck of GPU memory limitations that restrict AlphaFold 3-like models to processing only a few thousand residue...
-
Open Biomedical Knowledge Graphs at Scale: Construction, Federation, and AI Agent Access with Samyama Graph Database
This paper addresses the core pain point of fragmented biomedical data by constructing and federating large-scale, open knowledge graphs to enable sea...
-
Predictive Analytics for Foot Ulcers Using Time-Series Temperature and Pressure Data
This paper addresses the critical need for continuous, real-time monitoring of diabetic foot health by developing an unsupervised anomaly detection fr...
-
Hypothesis-Based Particle Detection for Accurate Nanoparticle Counting and Digital Diagnostics
This paper addresses the core challenge of achieving accurate, interpretable, and training-free nanoparticle counting in digital diagnostic assays, wh...
Contrastive Deep Learning for Variant Detection in Wastewater Genomic Sequencing
Georgia State University, Atlanta, Georgia, USA
30秒速读
IN SHORT: This paper addresses the core challenge of detecting viral variants in wastewater sequencing data without reference genomes or labeled annotations, overcoming issues of high noise, low coverage, and fragmented reads.
核心创新
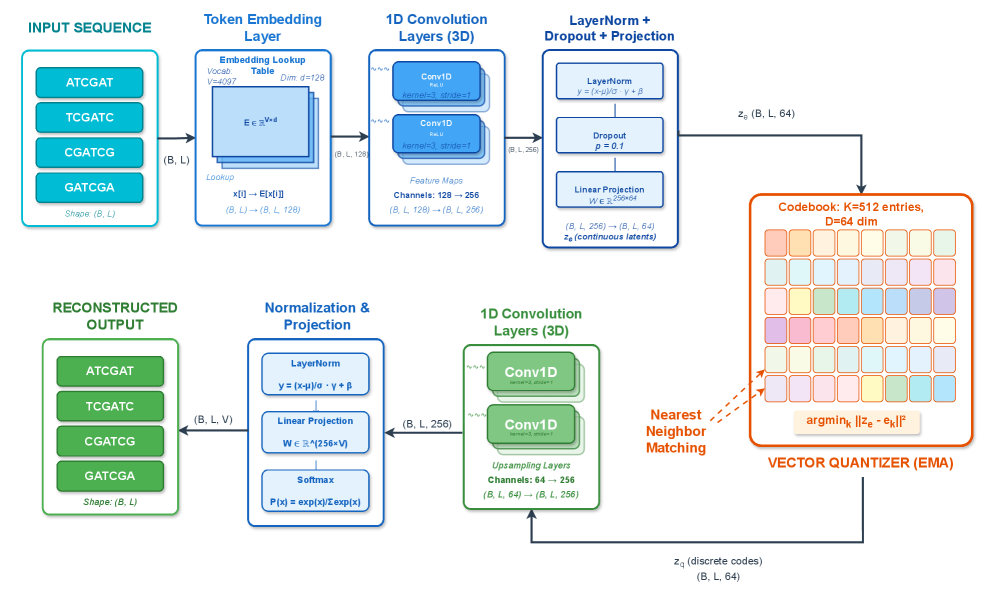
- Methodology First comprehensive application of VQ-VAE with EMA quantization to wastewater genomic surveillance, achieving 99.52% token-level reconstruction accuracy with 19.73% codebook utilization.
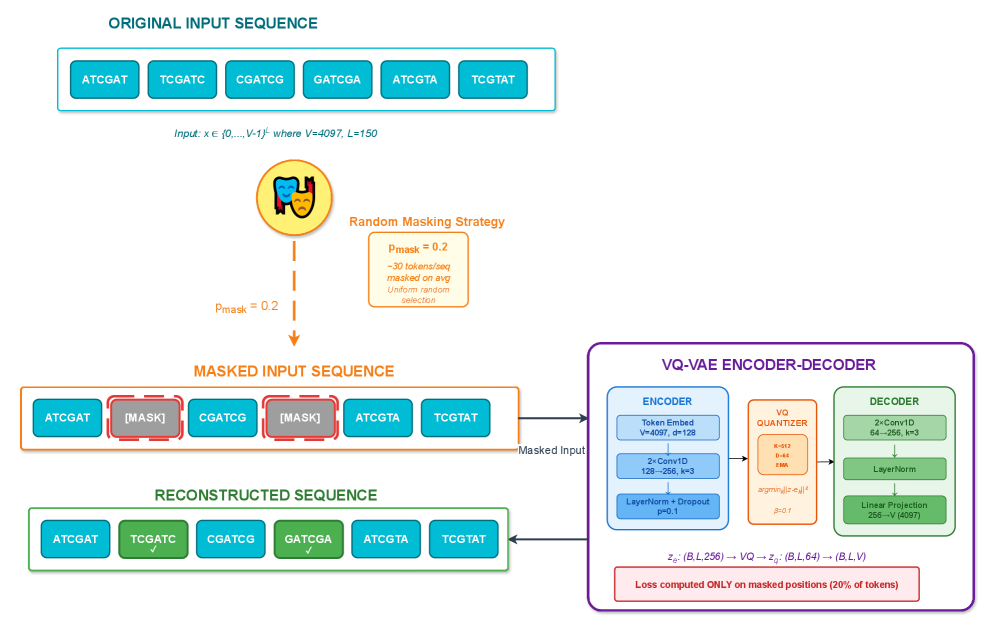
- Methodology Integration of masked reconstruction pretraining (BERT-style) maintaining ~95% accuracy under 20% token corruption, enabling robust inference with missing/low-quality data.
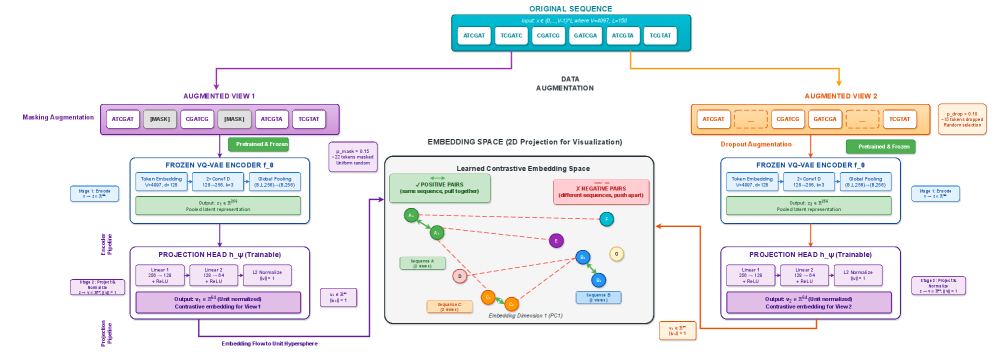
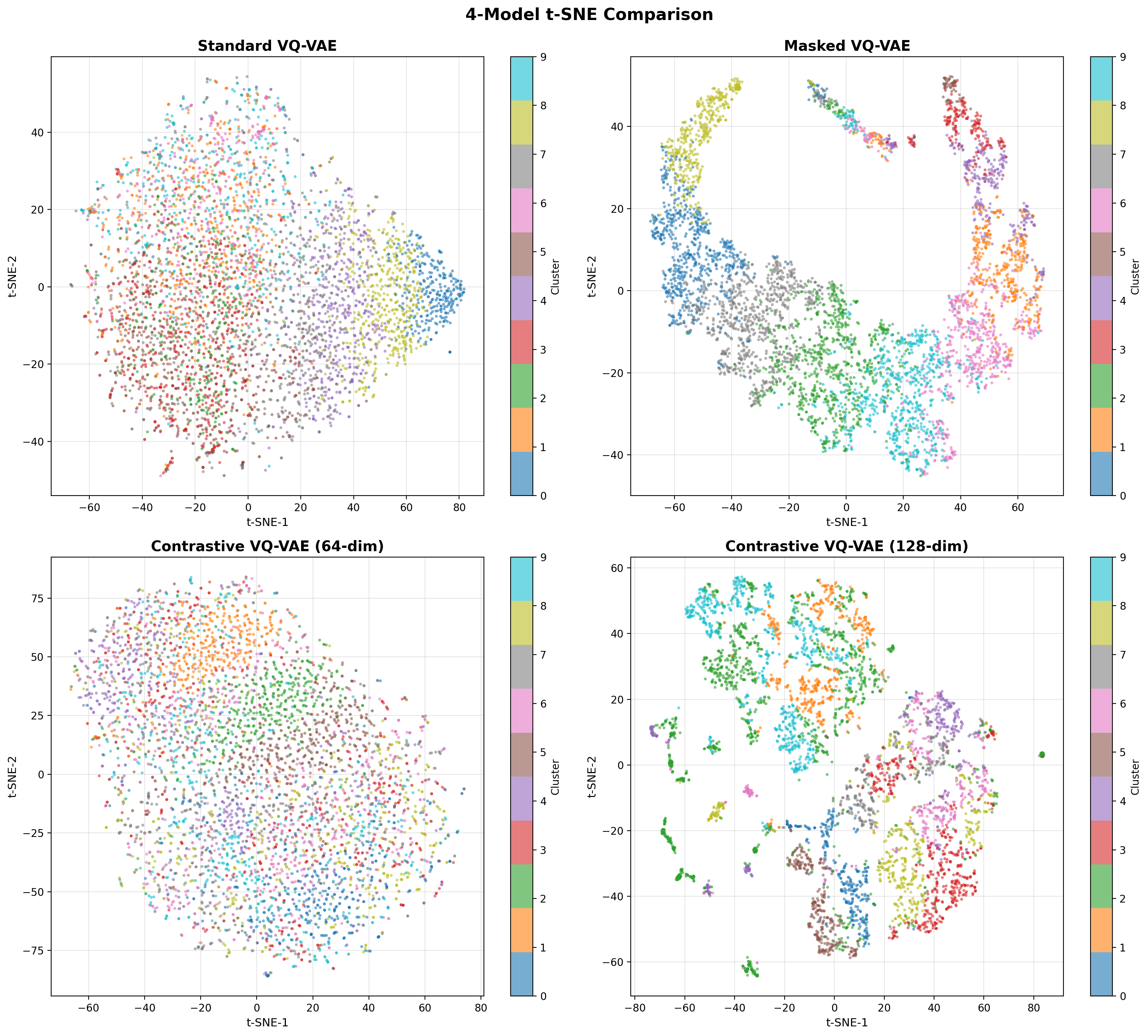
- Methodology Contrastive fine-tuning with varying embedding dimensions showing +35% (64-dim) and +42% (128-dim) Silhouette score improvements, establishing representation capacity impact on variant discrimination.
主要结论
- VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate on SARS-CoV-2 wastewater data with 100,000 reads.
- Contrastive fine-tuning improves clustering performance by +35% (0.31→0.42) with 64-dim embeddings and +42% (0.31→0.44) with 128-dim embeddings.
- The framework maintains efficient codebook utilization (19.73%, 101 of 512 codes active) while providing robust performance under data corruption.
摘要: Wastewater-based genomic surveillance has emerged as a powerful tool for population-level viral monitoring, offering comprehensive insights into circulating viral variants across entire communities. However, this approach faces significant computational challenges stemming from high sequencing noise, low viral coverage, fragmented reads, and the complete absence of labeled variant annotations. Traditional reference-based variant calling pipelines struggle with novel mutations and require extensive computational resources. We present a comprehensive framework for unsupervised viral variant detection using Vector-Quantized Variational Autoencoders (VQ-VAE) that learns discrete codebooks of genomic patterns from k-mer tokenized sequences without requiring reference genomes or variant labels. Our approach extends the base VQ-VAE architecture with masked reconstruction pretraining for robustness to missing data and contrastive learning for highly discriminative embeddings. Evaluated on SARS-CoV-2 wastewater sequencing data comprising approximately 100,000 reads, our VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate while maintaining 19.73% codebook utilization (101 of 512 codes active), demonstrating efficient discrete representation learning. Contrastive fine-tuning with different projection dimensions yields substantial clustering improvements: 64-dimensional embeddings achieve +35% Silhouette score improvement (0.31→0.42), while 128-dimensional embeddings achieve +42% improvement (0.31→0.44), clearly demonstrating the impact of embedding dimensionality on variant discrimination capability. Our reference-free framework provides a scalable, interpretable approach to genomic surveillance with direct applications to public health monitoring.