
Paper List
-
Formation of Artificial Neural Assemblies by Biologically Plausible Inhibition Mechanisms
This work addresses the core limitation of the Assembly Calculus model—its fixed-size, biologically implausible k-WTA selection process—by introducing...
-
How to make the most of your masked language model for protein engineering
This paper addresses the critical bottleneck of efficiently sampling high-quality, diverse protein sequences from Masked Language Models (MLMs) for pr...
-
Module control in youth symptom networks across COVID-19
This paper addresses the core challenge of distinguishing whether a prolonged societal stressor (COVID-19) fundamentally reorganizes the architecture ...
-
JEDI: Jointly Embedded Inference of Neural Dynamics
This paper addresses the core challenge of inferring context-dependent neural dynamics from noisy, high-dimensional recordings using a single unified ...
-
ATP Level and Phosphorylation Free Energy Regulate Trigger-Wave Speed and Critical Nucleus Size in Cellular Biochemical Systems
This work addresses the core challenge of quantitatively predicting how the cellular energy state (ATP level and phosphorylation free energy) governs ...
-
Packaging Jupyter notebooks as installable desktop apps using LabConstrictor
This paper addresses the core pain point of ensuring Jupyter notebook reproducibility and accessibility across different computing environments, parti...
-
SNPgen: Phenotype-Supervised Genotype Representation and Synthetic Data Generation via Latent Diffusion
This paper addresses the core challenge of generating privacy-preserving synthetic genotype data that maintains both statistical fidelity and downstre...
-
Continuous Diffusion Transformers for Designing Synthetic Regulatory Elements
This paper addresses the challenge of efficiently generating novel, cell-type-specific regulatory DNA sequences with high predicted activity while min...
Contrastive Deep Learning for Variant Detection in Wastewater Genomic Sequencing
Georgia State University, Atlanta, Georgia, USA
30秒速读
IN SHORT: This paper addresses the core challenge of detecting viral variants in wastewater sequencing data without reference genomes or labeled annotations, overcoming issues of high noise, low coverage, and fragmented reads.
核心创新
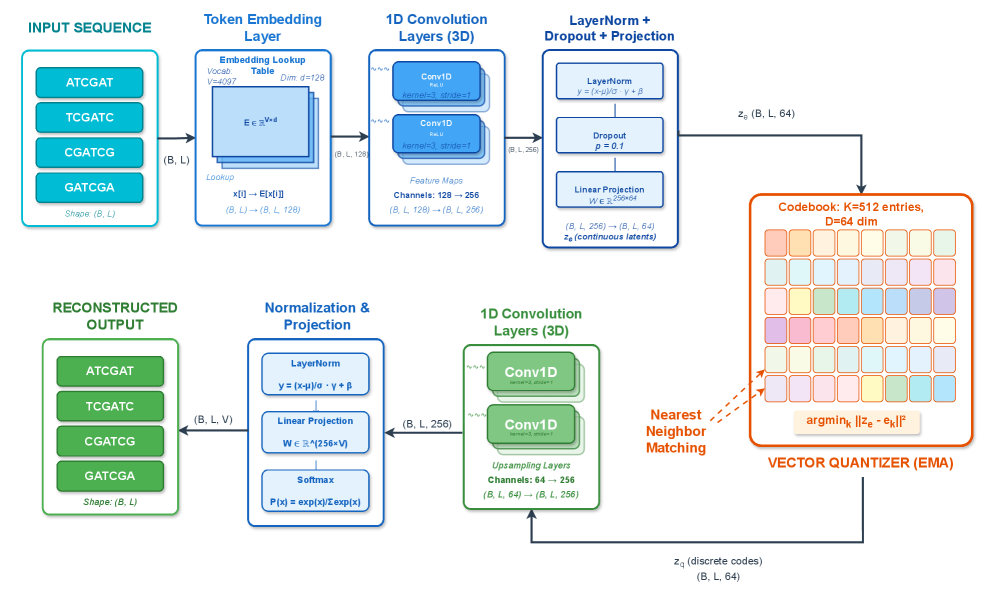
- Methodology First comprehensive application of VQ-VAE with EMA quantization to wastewater genomic surveillance, achieving 99.52% token-level reconstruction accuracy with 19.73% codebook utilization.
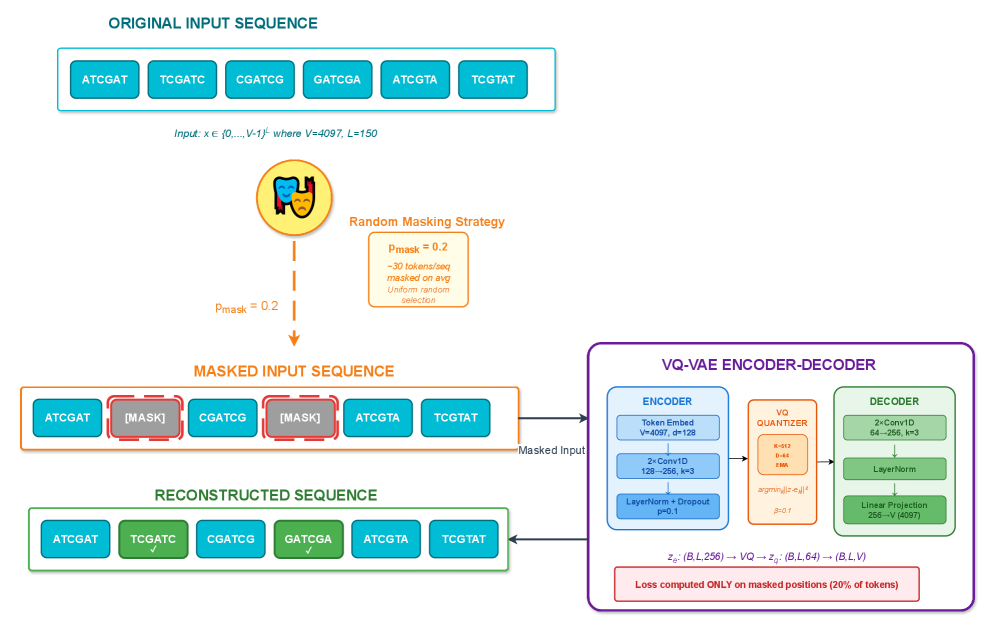
- Methodology Integration of masked reconstruction pretraining (BERT-style) maintaining ~95% accuracy under 20% token corruption, enabling robust inference with missing/low-quality data.
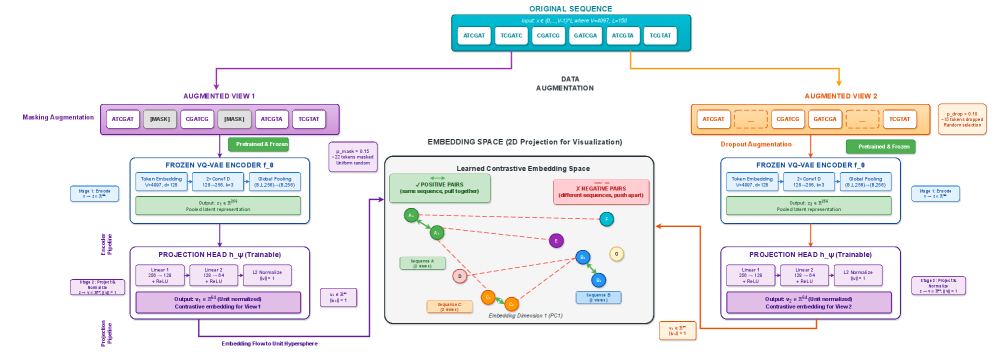
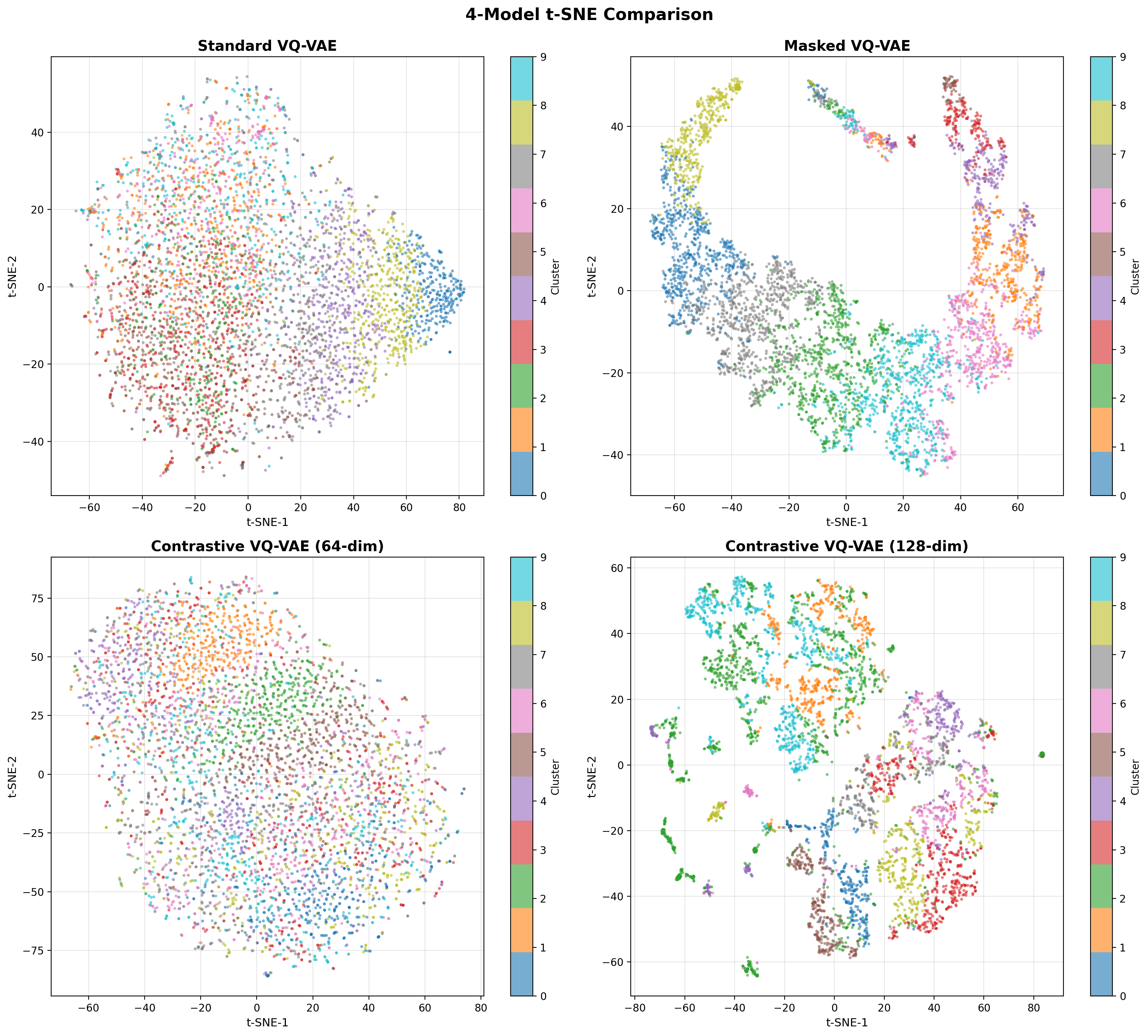
- Methodology Contrastive fine-tuning with varying embedding dimensions showing +35% (64-dim) and +42% (128-dim) Silhouette score improvements, establishing representation capacity impact on variant discrimination.
主要结论
- VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate on SARS-CoV-2 wastewater data with 100,000 reads.
- Contrastive fine-tuning improves clustering performance by +35% (0.31→0.42) with 64-dim embeddings and +42% (0.31→0.44) with 128-dim embeddings.
- The framework maintains efficient codebook utilization (19.73%, 101 of 512 codes active) while providing robust performance under data corruption.
摘要: Wastewater-based genomic surveillance has emerged as a powerful tool for population-level viral monitoring, offering comprehensive insights into circulating viral variants across entire communities. However, this approach faces significant computational challenges stemming from high sequencing noise, low viral coverage, fragmented reads, and the complete absence of labeled variant annotations. Traditional reference-based variant calling pipelines struggle with novel mutations and require extensive computational resources. We present a comprehensive framework for unsupervised viral variant detection using Vector-Quantized Variational Autoencoders (VQ-VAE) that learns discrete codebooks of genomic patterns from k-mer tokenized sequences without requiring reference genomes or variant labels. Our approach extends the base VQ-VAE architecture with masked reconstruction pretraining for robustness to missing data and contrastive learning for highly discriminative embeddings. Evaluated on SARS-CoV-2 wastewater sequencing data comprising approximately 100,000 reads, our VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate while maintaining 19.73% codebook utilization (101 of 512 codes active), demonstrating efficient discrete representation learning. Contrastive fine-tuning with different projection dimensions yields substantial clustering improvements: 64-dimensional embeddings achieve +35% Silhouette score improvement (0.31→0.42), while 128-dimensional embeddings achieve +42% improvement (0.31→0.44), clearly demonstrating the impact of embedding dimensionality on variant discrimination capability. Our reference-free framework provides a scalable, interpretable approach to genomic surveillance with direct applications to public health monitoring.