
Paper List
-
Discovery of a Hematopoietic Manifold in scGPT Yields a Method for Extracting Performant Algorithms from Biological Foundation Model Internals
This work addresses the core challenge of extracting reusable, interpretable, and high-performance biological algorithms from the opaque internal repr...
-
MS2MetGAN: Latent-space adversarial training for metabolite–spectrum matching in MS/MS database search
This paper addresses the critical bottleneck in metabolite identification: the generation of high-quality negative training samples that are structura...
-
Toward Robust, Reproducible, and Widely Accessible Intracranial Language Brain-Computer Interfaces: A Comprehensive Review of Neural Mechanisms, Hardware, Algorithms, Evaluation, Clinical Pathways and Future Directions
This review addresses the core challenge of fragmented and heterogeneous evidence that hinders the clinical translation of intracranial language BCIs,...
-
Less Is More in Chemotherapy of Breast Cancer
通过纳入细胞周期时滞和竞争项,解决了现有肿瘤-免疫模型的过度简化问题,以定量比较化疗方案。
-
Fold-CP: A Context Parallelism Framework for Biomolecular Modeling
This paper addresses the critical bottleneck of GPU memory limitations that restrict AlphaFold 3-like models to processing only a few thousand residue...
-
Open Biomedical Knowledge Graphs at Scale: Construction, Federation, and AI Agent Access with Samyama Graph Database
This paper addresses the core pain point of fragmented biomedical data by constructing and federating large-scale, open knowledge graphs to enable sea...
-
Predictive Analytics for Foot Ulcers Using Time-Series Temperature and Pressure Data
This paper addresses the critical need for continuous, real-time monitoring of diabetic foot health by developing an unsupervised anomaly detection fr...
-
Hypothesis-Based Particle Detection for Accurate Nanoparticle Counting and Digital Diagnostics
This paper addresses the core challenge of achieving accurate, interpretable, and training-free nanoparticle counting in digital diagnostic assays, wh...
PanFoMa: A Lightweight Foundation Model and Benchmark for Pan-Cancer
Extracted from affiliations in the content snippet (specific institutions not fully listed in provided text)

30秒速读
IN SHORT: This paper addresses the dual challenge of achieving computational efficiency without sacrificing accuracy in whole-transcriptome single-cell representation learning for pan-cancer analysis, moving beyond the limitations of pure Transformer or Mamba architectures.
核心创新
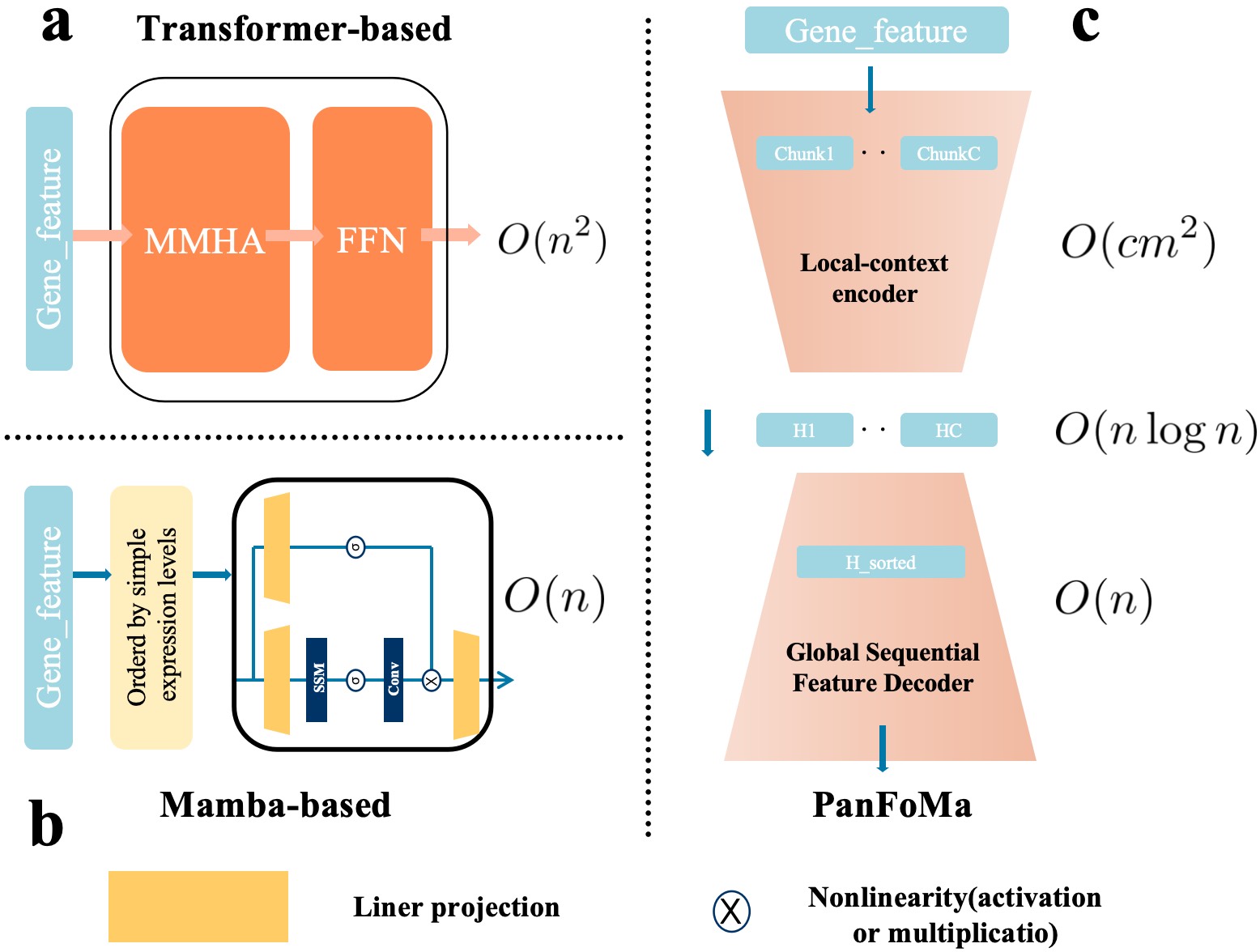
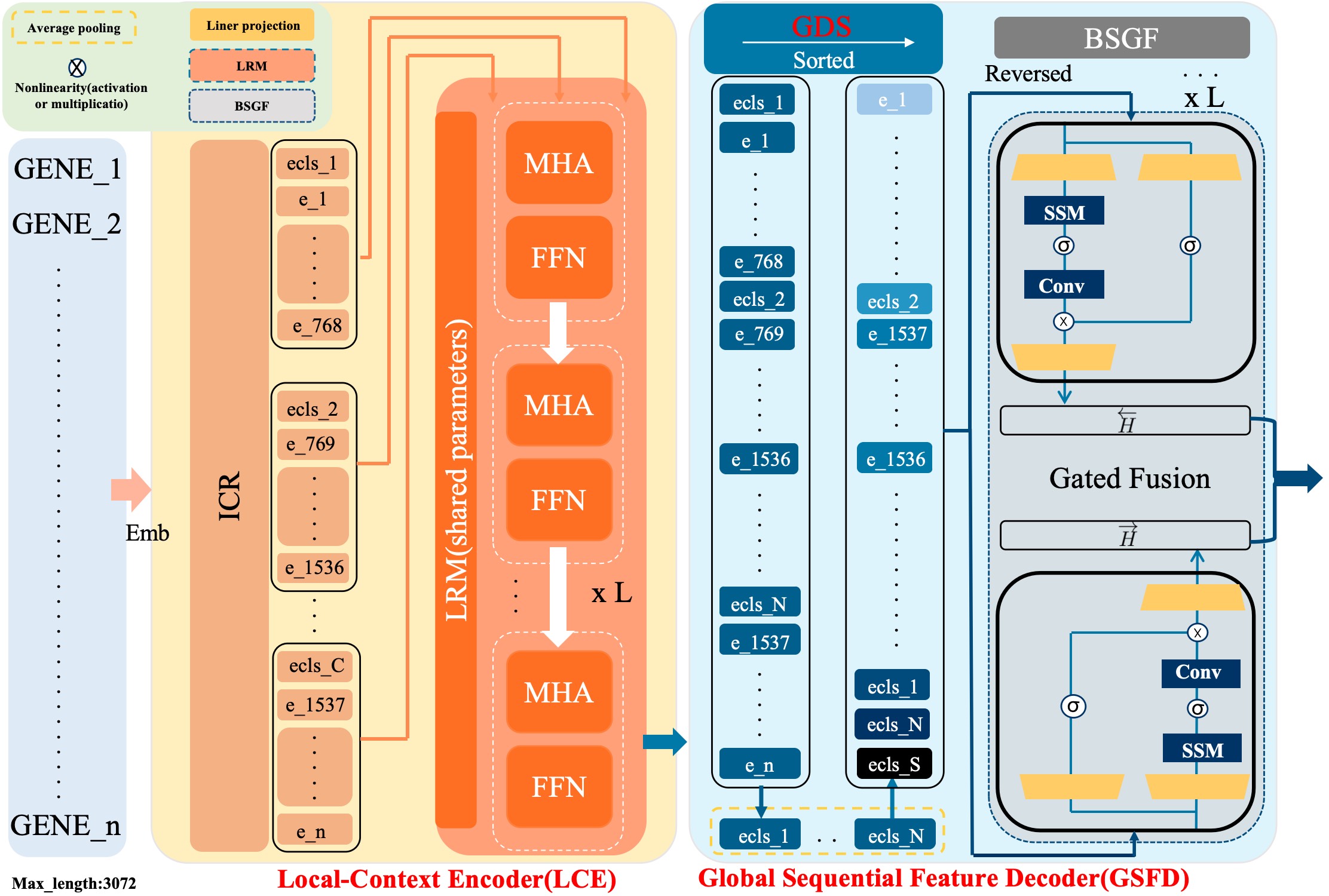
- Methodology Proposes a novel hybrid architecture (PanFoMa) that decouples local gene interaction modeling (via a lightweight, chunked Transformer encoder) from global context integration (via a bidirectional Mamba decoder), achieving O(C·M² + N log N) complexity.
- Methodology Introduces a Global-informed Dynamic Sorting (GDS) mechanism that adaptively orders genes for the Mamba decoder based on a learned global cell state vector, moving beyond static, heuristic gene ordering (e.g., by mean expression).
- Biology Constructs and releases PanFoMaBench, a large-scale, rigorously curated pan-cancer single-cell benchmark comprising over 3.5 million high-quality cells across 33 cancer subtypes from 23 tissues, addressing the lack of comprehensive evaluation resources.
主要结论
- PanFoMa achieves state-of-the-art pan-cancer classification accuracy of 94.74% (ACC) and 92.5% (Macro-F1) on PanFoMaBench, outperforming GeneFormer by +3.5% ACC and +4.0% F1.
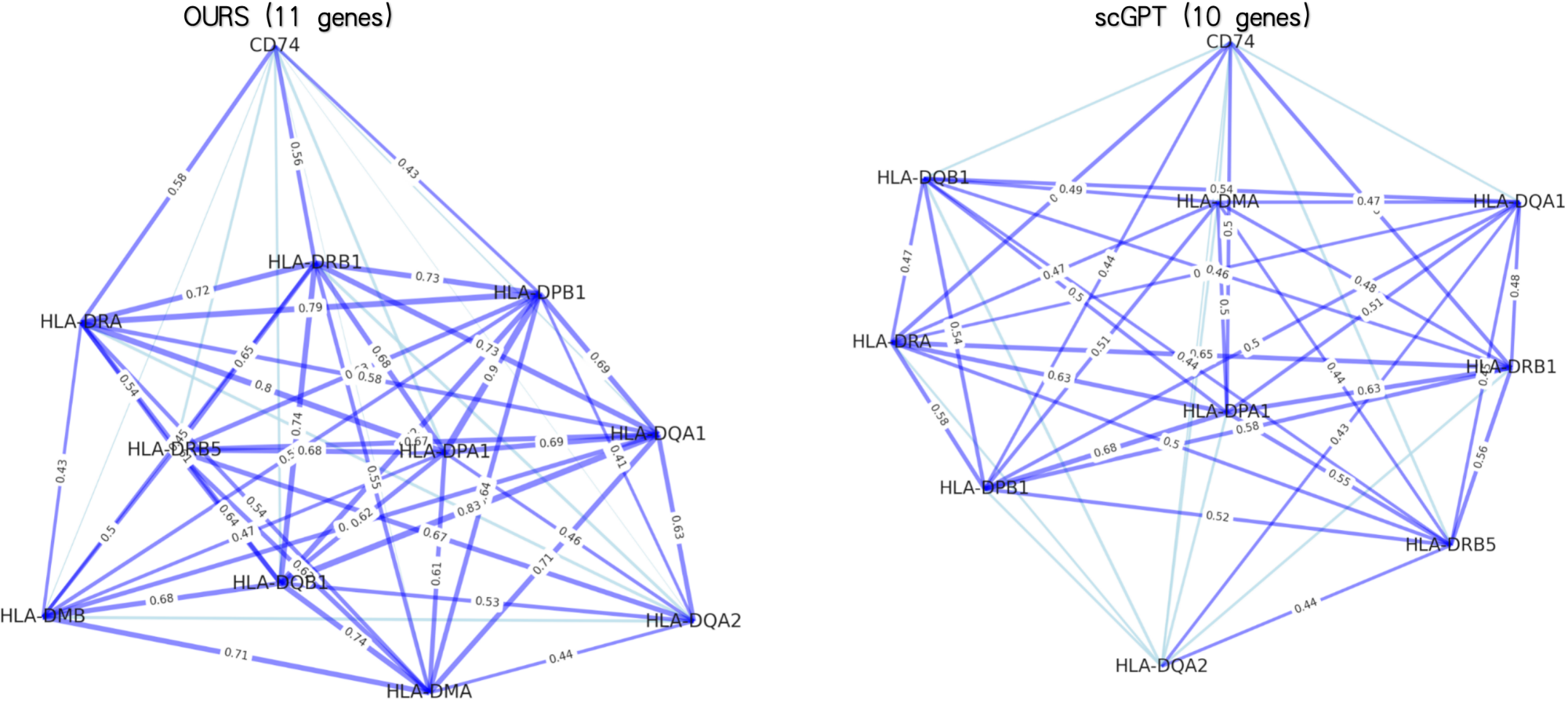
- The model demonstrates superior generalizability across foundational tasks, showing improvements of +7.4% in cell type annotation, +4.0% in batch integration, and +3.1% in multi-omics integration over baselines.
- The hybrid local-global design and dynamic sorting are validated as effective, enabling efficient processing of full transcriptome-scale data (~3000 genes) while capturing both fine-grained local interactions and broad global regulatory patterns.
摘要: Single-cell RNA sequencing (scRNA-seq) is essential for decoding tumor heterogeneity. However, pan-cancer research still faces two key challenges: learning discriminative and efficient single-cell representations, and establishing a comprehensive evaluation benchmark. In this paper, we introduce PanFoMa, a lightweight hybrid neural network that combines the strengths of Transformers and state-space models to achieve a balance between performance and efficiency. PanFoMa consists of a front-end local-context encoder with shared self-attention layers to capture complex, order-independent gene interactions; and a back-end global sequential feature decoder that efficiently integrates global context using a linear-time state-space model. This modular design preserves the expressive power of Transformers while leveraging the scalability of Mamba to enable transcriptome modeling, effectively capturing both local and global regulatory signals. To enable robust evaluation, we also construct a large-scale pan-cancer single-cell benchmark, PanFoMaBench, containing over 3.5 million high-quality cells across 33 cancer subtypes, curated through a rigorous preprocessing pipeline. Experimental results show that PanFoMa outperforms state-of-the-art models on our pan-cancer benchmark (+4.0%) and across multiple public tasks, including cell type annotation (+7.4%), batch integration (+4.0%) and multi-omics integration (+3.1%). The code is available at https://github.com/Xiaoshui-Huang/PanFoMa.