
Paper List
-
Discovery of a Hematopoietic Manifold in scGPT Yields a Method for Extracting Performant Algorithms from Biological Foundation Model Internals
This work addresses the core challenge of extracting reusable, interpretable, and high-performance biological algorithms from the opaque internal repr...
-
MS2MetGAN: Latent-space adversarial training for metabolite–spectrum matching in MS/MS database search
This paper addresses the critical bottleneck in metabolite identification: the generation of high-quality negative training samples that are structura...
-
Toward Robust, Reproducible, and Widely Accessible Intracranial Language Brain-Computer Interfaces: A Comprehensive Review of Neural Mechanisms, Hardware, Algorithms, Evaluation, Clinical Pathways and Future Directions
This review addresses the core challenge of fragmented and heterogeneous evidence that hinders the clinical translation of intracranial language BCIs,...
-
Less Is More in Chemotherapy of Breast Cancer
通过纳入细胞周期时滞和竞争项,解决了现有肿瘤-免疫模型的过度简化问题,以定量比较化疗方案。
-
Fold-CP: A Context Parallelism Framework for Biomolecular Modeling
This paper addresses the critical bottleneck of GPU memory limitations that restrict AlphaFold 3-like models to processing only a few thousand residue...
-
Open Biomedical Knowledge Graphs at Scale: Construction, Federation, and AI Agent Access with Samyama Graph Database
This paper addresses the core pain point of fragmented biomedical data by constructing and federating large-scale, open knowledge graphs to enable sea...
-
Predictive Analytics for Foot Ulcers Using Time-Series Temperature and Pressure Data
This paper addresses the critical need for continuous, real-time monitoring of diabetic foot health by developing an unsupervised anomaly detection fr...
-
Hypothesis-Based Particle Detection for Accurate Nanoparticle Counting and Digital Diagnostics
This paper addresses the core challenge of achieving accurate, interpretable, and training-free nanoparticle counting in digital diagnostic assays, wh...
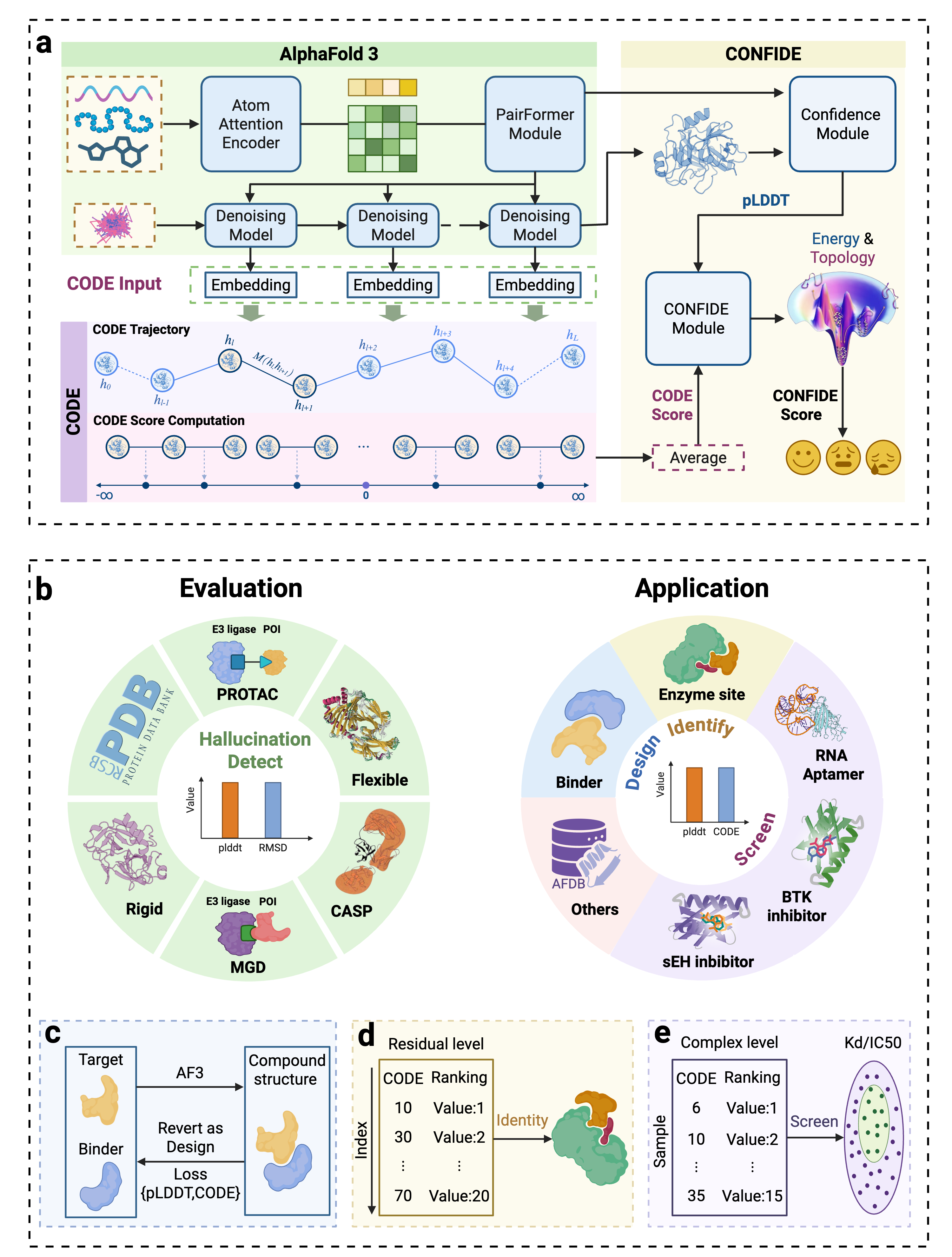
CONFIDE: Hallucination Assessment for Reliable Biomolecular Structure Prediction and Design
The Chinese University of Hong Kong | Zhejiang University | Macao Polytechnic University | University of Electronic Science and Technology of China
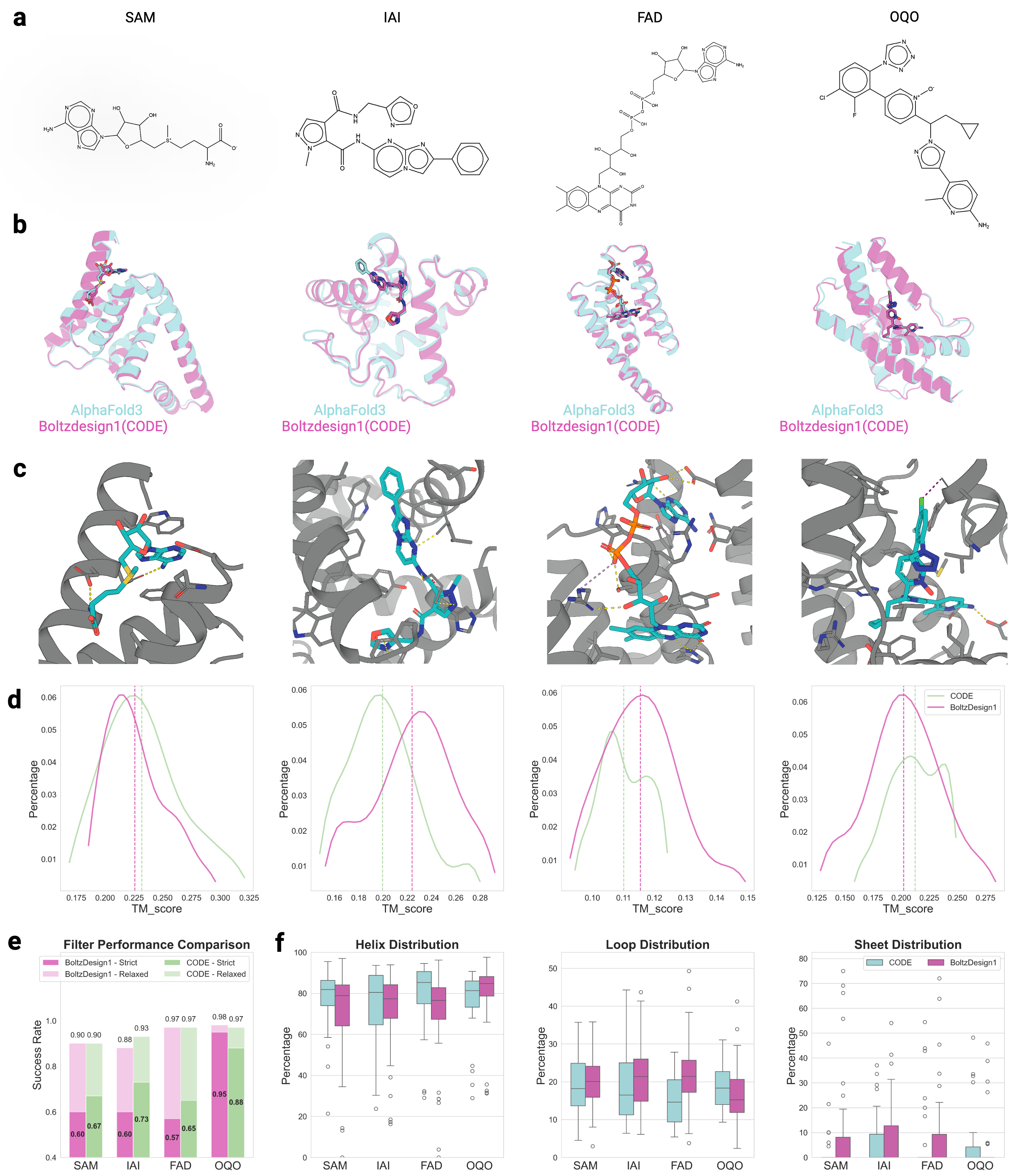
30秒速读
IN SHORT: This paper addresses the critical limitation of current protein structure prediction models (like AlphaFold3) where high-confidence scores (pLDDT) can be misleading, failing to detect subtle structural errors like atomic clashes and topological traps, which undermines reliability in downstream applications like drug discovery.
核心创新
- Methodology Introduces CODE (Chain of Diffusion Embeddings), a novel, unsupervised metric derived from AlphaFold3's latent diffusion embeddings that directly quantifies topological frustration, a key factor in protein folding kinetics previously overlooked by confidence scores.
- Methodology Proposes CONFIDE, a unified evaluation framework that integrates the energetic perspective of pLDDT with the topological perspective of CODE, providing a more comprehensive and reliable assessment of predicted biomolecular structures.
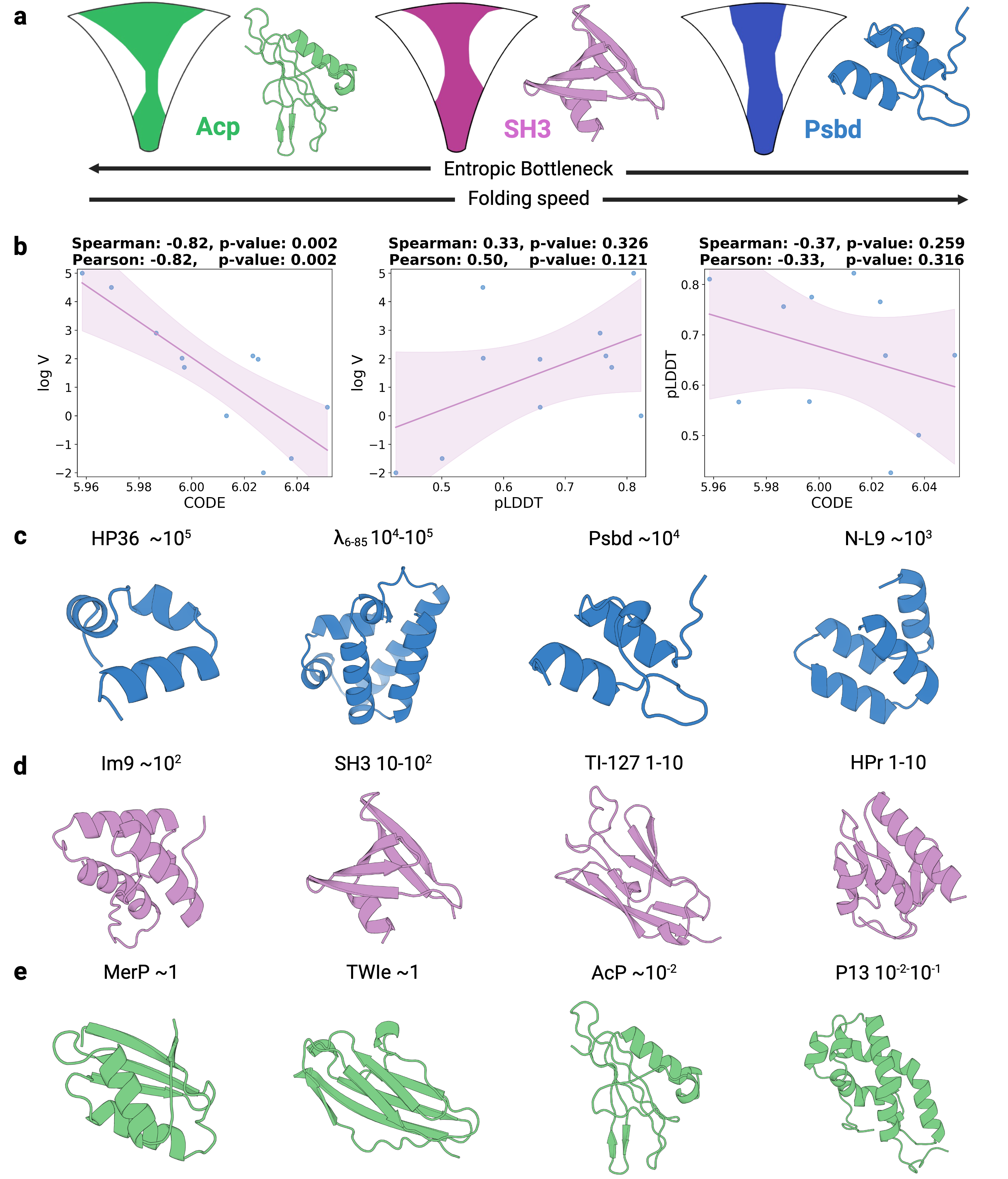
- Biology Establishes a strong empirical link between the CODE metric and protein folding rates driven by topological frustration (Spearman correlation of -0.82, p=0.002), offering a data-driven proxy for a complex biophysical phenomenon.
主要结论
- CODE demonstrates a strong, statistically significant correlation with protein folding rates mediated by topological frustration (Spearman ρ = -0.82, p=0.002), far outperforming pLDDT (ρ = 0.33, p=0.326).
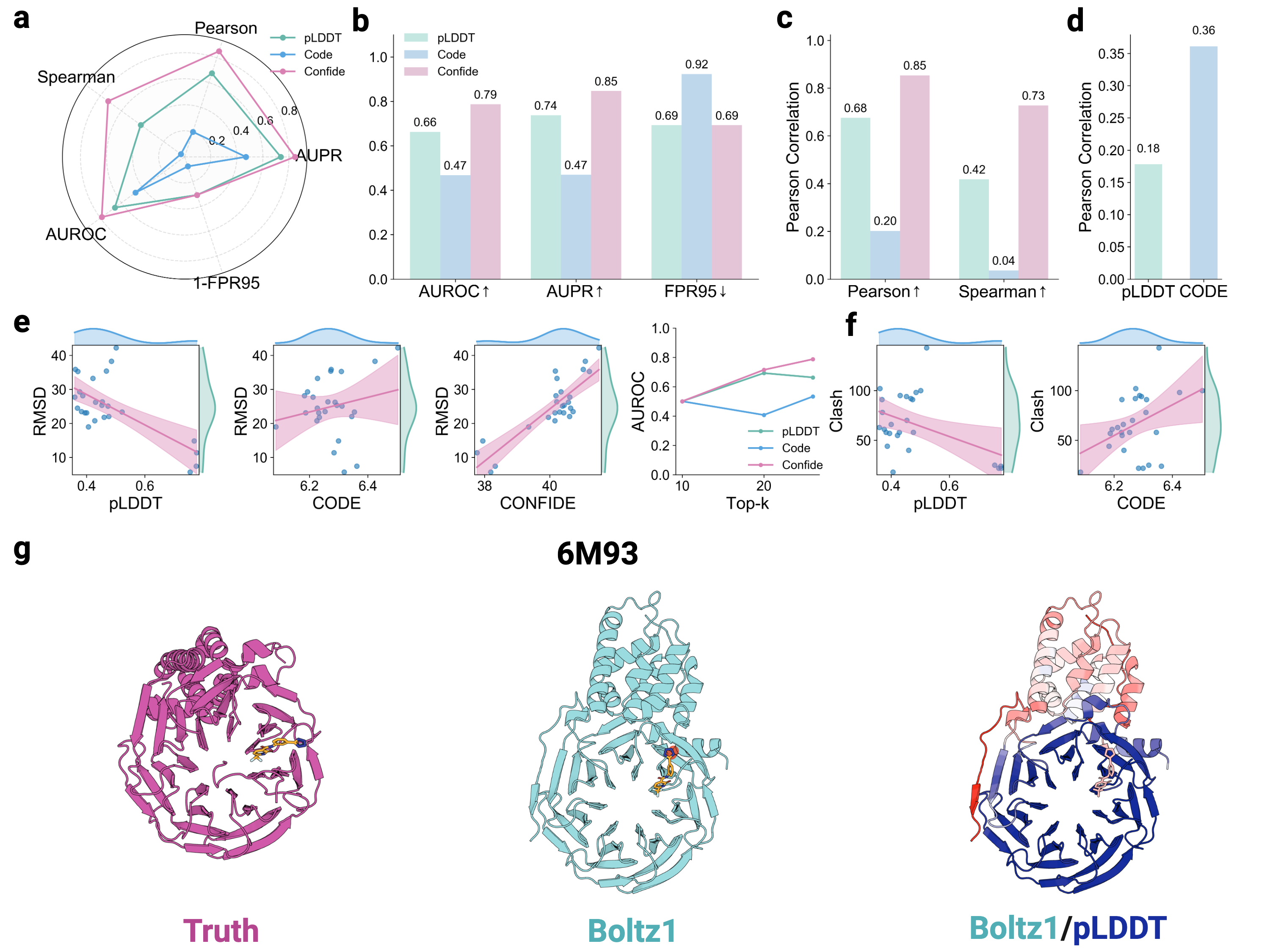
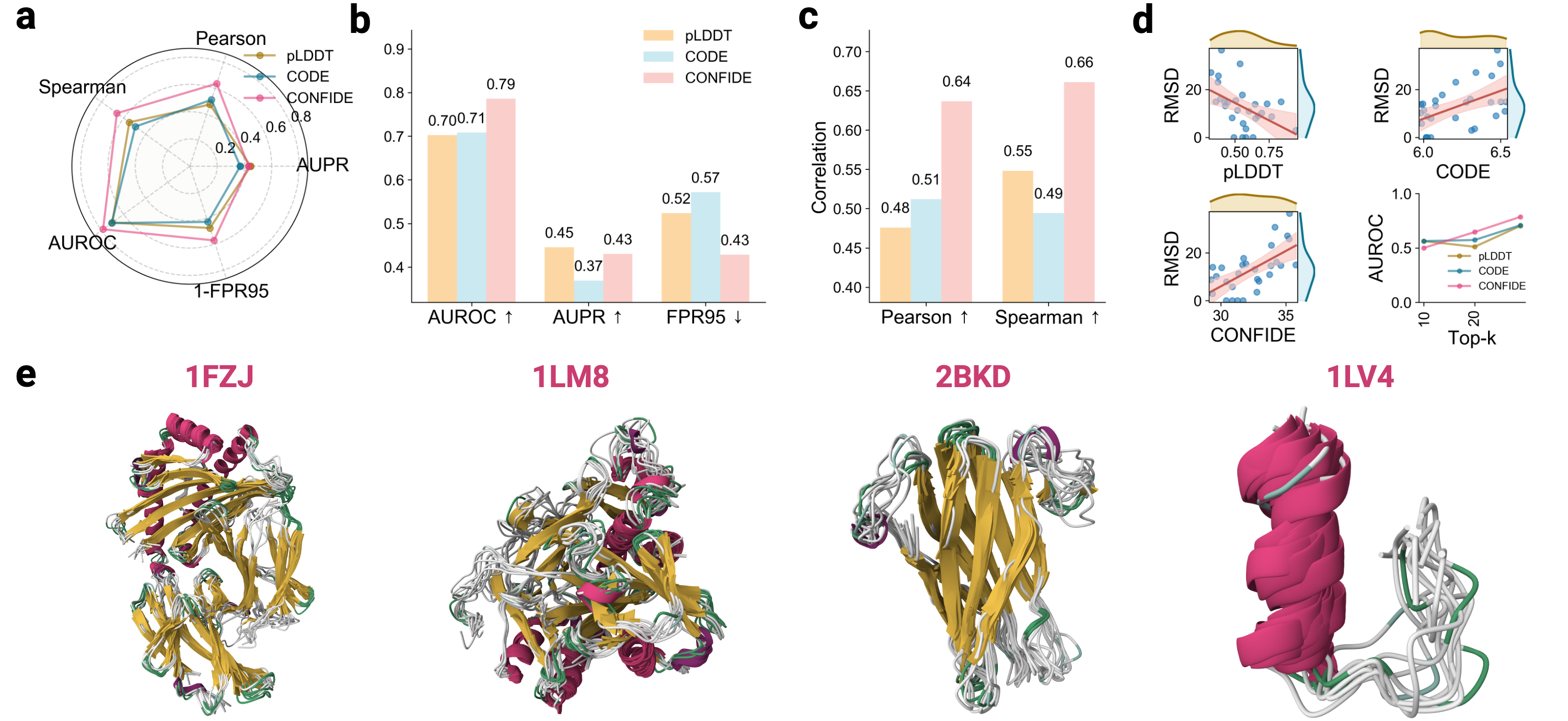
- The CONFIDE framework significantly improves hallucination detection, achieving a Spearman correlation of 0.73 with RMSD on molecular glue benchmarks, a 73.8% relative improvement over pLDDT's correlation of 0.42.
- CONFIDE enables practical downstream applications, improving binder design success rates (e.g., +13% for IAI) and accurately predicting mutation-induced binding affinity changes (Spearman ρ = 0.83 for BTK vs. Fenebrutinib, compared to pLDDT's ρ = 0.03).
摘要: Reliable evaluation of protein structure predictions remains challenging, as metrics like pLDDT capture energetic stability but often miss subtle errors such as atomic clashes or conformational traps reflecting topological frustration within the protein-folding energy landscape. We present CODE (Chain of Diffusion Embeddings), a self-evaluating metric empirically found to quantify topological frustration directly from the latent diffusion embeddings of the AlphaFold3 series of structure predictors in a fully unsupervised manner. Integrating this with pLDDT, we propose CONFIDE, a unified evaluation framework that combines energetic and topological perspectives to improve the reliability of AlphaFold3 and related models. CODE strongly correlates with protein folding rates driven by topological frustration, achieving a correlation of 0.82 compared to pLDDT’s 0.33 (a relative improvement of 148%). CONFIDE significantly enhances the reliability of quality evaluation in molecular glue structure prediction benchmarks, achieving a Spearman correlation of 0.73 with RMSD, compared to pLDDT’s correlation of 0.42, a relative improvement of 73.8%. Beyond quality assessment, our approach applies to diverse drug-design tasks, including all-atom binder design, enzymatic active-site mapping, mutation-induced binding-affinity prediction, nucleic acid aptamer screening, and flexible protein modeling. By combining data-driven embeddings with theoretical insight, CODE and CONFIDE outperform existing metrics across a wide range of biomolecular systems, offering robust and versatile tools to refine structure predictions, advance structural biology, and accelerate drug discovery.