
Paper List
-
Developing the PsyCogMetrics™ AI Lab to Evaluate Large Language Models and Advance Cognitive Science
This paper addresses the critical gap between sophisticated LLM evaluation needs and the lack of accessible, scientifically rigorous platforms that in...
-
Equivalence of approximation by networks of single- and multi-spike neurons
This paper resolves the fundamental question of whether single-spike spiking neural networks (SNNs) are inherently less expressive than multi-spike SN...
-
The neuroscience of transformers
提出了Transformer架构与皮层柱微环路之间的新颖计算映射,连接了现代AI与神经科学。
-
Framing local structural identifiability and observability in terms of parameter-state symmetries
This paper addresses the core challenge of systematically determining which parameters and states in a mechanistic ODE model can be uniquely inferred ...
-
Leveraging Phytolith Research using Artificial Intelligence
This paper addresses the critical bottleneck in phytolith research by automating the labor-intensive manual microscopy process through a multimodal AI...
-
Neural network-based encoding in free-viewing fMRI with gaze-aware models
This paper addresses the core challenge of building computationally efficient and ecologically valid brain encoding models for naturalistic vision by ...
-
Scalable DNA Ternary Full Adder Enabled by a Competitive Blocking Circuit
This paper addresses the core bottleneck of carry information attenuation and limited computational scale in DNA binary adders by introducing a scalab...
-
ELISA: An Interpretable Hybrid Generative AI Agent for Expression-Grounded Discovery in Single-Cell Genomics
This paper addresses the critical bottleneck of translating high-dimensional single-cell transcriptomic data into interpretable biological hypotheses ...
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
Department of Computer Science and Genome Center, University of California, Davis | Architecture et Dynamique des Macromolécules Biologiques, UMR 3528 du CNRS, Institut Pasteur | Department of Physics, School of Sciences, Great Bay University | Université Paris-Saclay, CNRS, CEA, Institut de Physique Théorique
30秒速读
IN SHORT: This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein conformations, a problem where existing methods often produce non-physical trajectories due to oversimplified energy surfaces and steric clashes.
核心创新
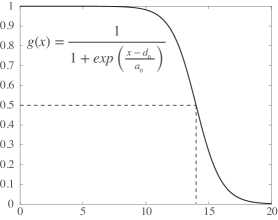
- Methodology Introduces SIDE (Stochastic Integro-Differential Equation), a novel Langevin bridge-based framework that efficiently approximates exact bridge equations at low temperatures to generate constrained transition trajectories.
- Methodology Develops a new coarse-grained potential that combines a Gō-like term (to preserve native backbone geometry) with a Rouse-type elastic energy term (from polymer physics), avoiding the problematic mixing of start/target conformation information used in prior methods like MinActionPath.
- Theory Provides a rigorous stochastic integro-differential formulation derived from the Langevin bridge formalism, which explicitly constrains trajectories to reach a target state within finite time, moving beyond Minimum Action Path (MAP) principles.
主要结论
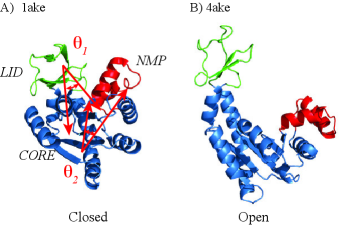
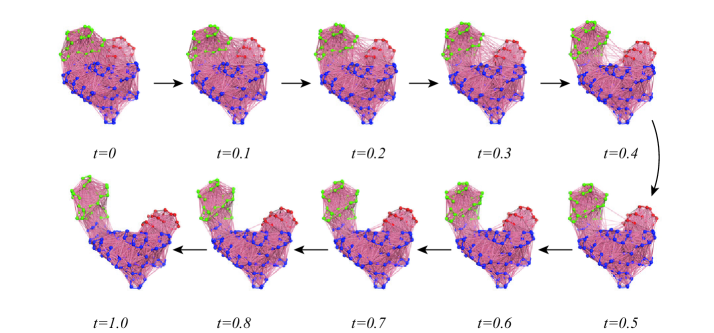
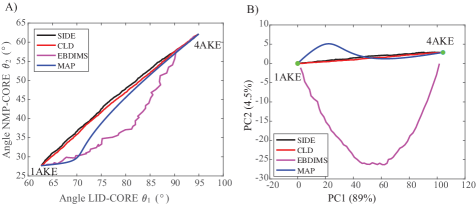
- The SIDE framework generates smooth, low-energy transition trajectories that maintain realistic molecular geometry, as demonstrated on several proteins undergoing large-scale conformational changes.
- SIDE frequently recovers experimentally supported intermediate states along transition paths, suggesting its paths have biological relevance beyond mere endpoint interpolation.
- Compared to established methods like MinActionPath and EBDIMS, SIDE offers improved physical realism and computational efficiency for modeling biomolecular conformational transitions, though challenges remain for highly complex motions.
摘要: We introduce a computational framework for generating realistic transition paths between distinct conformations of large biomolecular systems. The method is built on a stochastic integro-differential formulation derived from the Langevin bridge formalism, which constrains molecular trajectories to reach a prescribed final state within a finite time and yields an efficient low-temperature approximation of the exact bridge equation. To obtain physically meaningful protein transitions, we couple this formulation to a new coarse-grained potential combining a Gō-like term that preserves native backbone geometry with a Rouse-type elastic energy term from polymer physics; we refer to the resulting approach as SIDE. We evaluate SIDE on several proteins undergoing large-scale conformational changes and compare its performance with established methods such as MinActionPath and EBDIMS. SIDE generates smooth, low-energy trajectories that maintain molecular geometry and frequently recover experimentally supported intermediate states. Although challenges remain for highly complex motions—largely due to the simplified coarse-grained potential—our results demonstrate that SIDE offers a powerful and computationally efficient strategy for modeling biomolecular conformational transitions.