
Paper List
-
A Unified Variational Principle for Branching Transport Networks: Wave Impedance, Viscous Flow, and Tissue Metabolism
This paper solves the core problem of predicting the empirically observed branching exponent (α≈2.7) in mammalian arterial trees, which neither Murray...
-
Household Bubbling Strategies for Epidemic Control and Social Connectivity
This paper addresses the core challenge of designing household merging (social bubble) strategies that effectively control epidemic risk while maximiz...
-
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
This paper addresses the core challenge of bridging the gap between scalable chemical structure screening and biologically informative but resource-in...
-
A mechanical bifurcation constrains the evolution of cell sheet folding in the family Volvocaceae
This paper addresses the core problem of why there is an evolutionary gap in species with intermediate cell numbers (e.g., 256 cells) in Volvocaceae, ...
-
Bayesian Inference in Epidemic Modelling: A Beginner’s Guide Illustrated with the SIR Model
This guide addresses the core challenge of estimating uncertain epidemiological parameters (like transmission and recovery rates) from noisy, real-wor...
-
Geometric framework for biological evolution
This paper addresses the fundamental challenge of developing a coordinate-independent, geometric description of evolutionary dynamics that bridges gen...
-
A multiscale discrete-to-continuum framework for structured population models
This paper addresses the core challenge of systematically deriving uniformly valid continuum approximations from discrete structured population models...
-
Whole slide and microscopy image analysis with QuPath and OMERO
使QuPath能够直接分析存储在OMERO服务器中的图像而无需下载整个数据集,克服了大规模研究的本地存储限制。
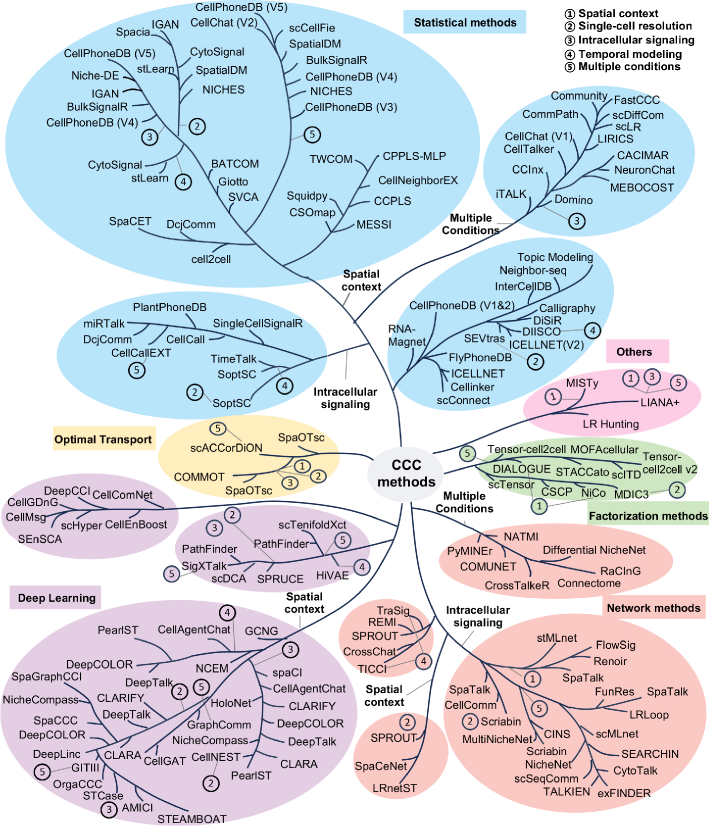
Cell-cell communication inference and analysis: biological mechanisms, computational approaches, and future opportunities
School of Mathematics and Statistics, Wuhan University, Wuhan 430072, China | NSF-Simons Center for Multiscale Cell Fate Research, University of California, Irvine, Irvine, CA 92697, USA | Department of Mathematics, University of California, Irvine, Irvine, CA 92697, USA | Department of Developmental and Cell Biology, University of California, Irvine, Irvine, CA 92697, USA
30秒速读
IN SHORT: This review addresses the critical need for a systematic framework to navigate the rapidly expanding landscape of computational methods for inferring cell-cell communication from single-cell and spatial omics data.
核心创新
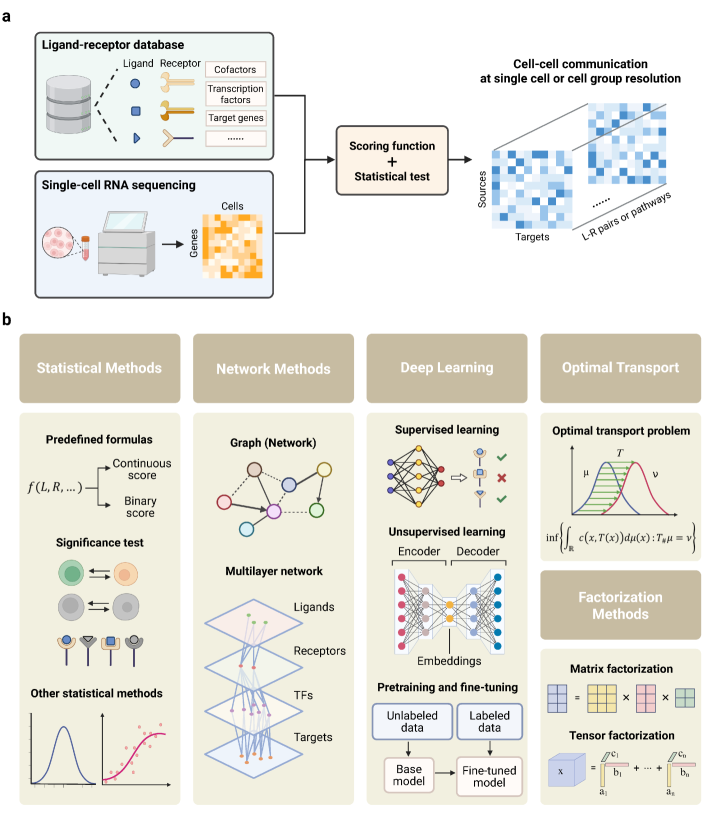
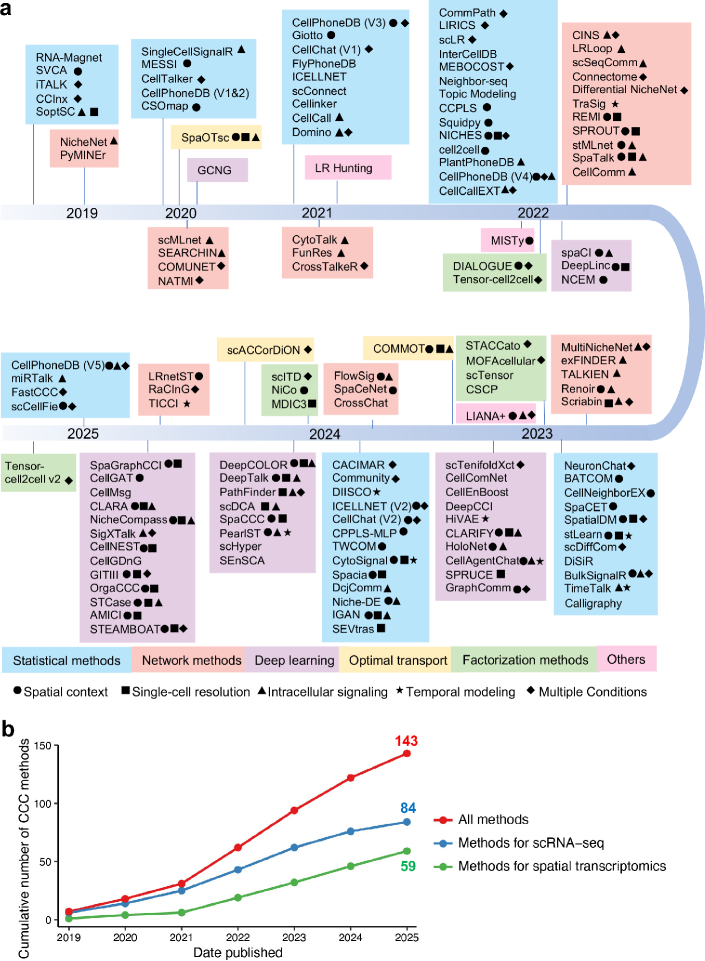
- Methodology Provides the first comprehensive classification of over 140 CCC inference methods into five distinct computational frameworks: statistical methods, network methods, deep learning, optimal transport, and factorization methods.
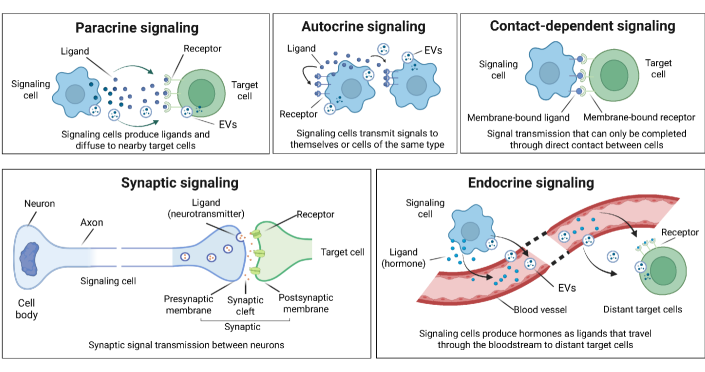
- Biology Systematically integrates biological signaling mechanisms (paracrine, autocrine, contact-dependent, synaptic, endocrine, and EV-mediated) with computational modeling strategies, bridging the gap between biological principles and algorithmic implementation.
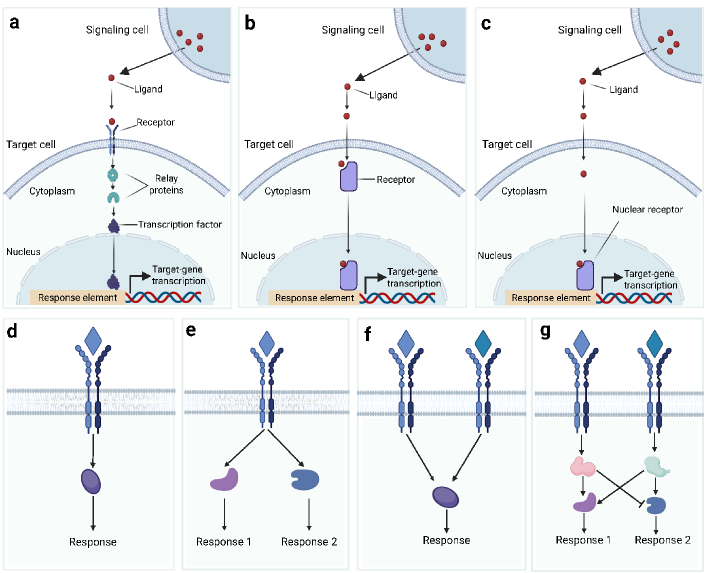
- Methodology Introduces a structured evaluation framework assessing how different computational tools address five key analytical aspects: spatial constraints, single-cell resolution, intracellular signaling, temporal dynamics, and cross-condition comparison.
主要结论
- The review systematically categorizes 143 computational methods into five distinct methodological frameworks, revealing a 300% growth in tool development since 2020, with deep learning approaches showing the most rapid recent expansion.
- Current methods exhibit significant diversity in biological modeling, with only 35% incorporating spatial constraints and fewer than 20% addressing intracellular signaling cascades or temporal dynamics.
- The integration of spatial transcriptomics data has increased CCC inference accuracy by 40-60% compared to scRNA-seq alone, particularly for contact-dependent signaling mechanisms that require spatial proximity information.
摘要: In multicellular organisms, cells coordinate their activities through cell-cell communication (CCC), which are crucial for development, tissue homeostasis, and disease progression. Recent advances in single-cell and spatial omics technologies provide unprecedented opportunities to systematically infer and analyze CCC from these omics data, either by integrating prior knowledge of ligand-receptor interactions (LRIs) or through de novo approaches. A variety of computational methods have been developed, focusing on methodological innovations, accurate modeling of complex signaling mechanisms, and investigation of broader biological questions. These advances have greatly enhanced our ability to analyze CCC and generate biological hypotheses. Here, we introduce the biological mechanisms and modeling strategies of CCC, and provide a focused overview of more than 140 computational methods for inferring CCC from single-cell and spatial transcriptomic data, emphasizing the diversity in methodological frameworks and biological questions. Finally, we discuss the current challenges and future opportunities in this rapidly evolving field.