
Paper List
-
Discovery of a Hematopoietic Manifold in scGPT Yields a Method for Extracting Performant Algorithms from Biological Foundation Model Internals
This work addresses the core challenge of extracting reusable, interpretable, and high-performance biological algorithms from the opaque internal repr...
-
MS2MetGAN: Latent-space adversarial training for metabolite–spectrum matching in MS/MS database search
This paper addresses the critical bottleneck in metabolite identification: the generation of high-quality negative training samples that are structura...
-
Toward Robust, Reproducible, and Widely Accessible Intracranial Language Brain-Computer Interfaces: A Comprehensive Review of Neural Mechanisms, Hardware, Algorithms, Evaluation, Clinical Pathways and Future Directions
This review addresses the core challenge of fragmented and heterogeneous evidence that hinders the clinical translation of intracranial language BCIs,...
-
Less Is More in Chemotherapy of Breast Cancer
通过纳入细胞周期时滞和竞争项,解决了现有肿瘤-免疫模型的过度简化问题,以定量比较化疗方案。
-
Fold-CP: A Context Parallelism Framework for Biomolecular Modeling
This paper addresses the critical bottleneck of GPU memory limitations that restrict AlphaFold 3-like models to processing only a few thousand residue...
-
Open Biomedical Knowledge Graphs at Scale: Construction, Federation, and AI Agent Access with Samyama Graph Database
This paper addresses the core pain point of fragmented biomedical data by constructing and federating large-scale, open knowledge graphs to enable sea...
-
Predictive Analytics for Foot Ulcers Using Time-Series Temperature and Pressure Data
This paper addresses the critical need for continuous, real-time monitoring of diabetic foot health by developing an unsupervised anomaly detection fr...
-
Hypothesis-Based Particle Detection for Accurate Nanoparticle Counting and Digital Diagnostics
This paper addresses the core challenge of achieving accurate, interpretable, and training-free nanoparticle counting in digital diagnostic assays, wh...
Unlocking hidden biomolecular conformational landscapes in diffusion models at inference time
Stanford University | Yale School of Medicine

30秒速读
IN SHORT: This paper addresses the core challenge of efficiently and accurately sampling the conformational landscape of biomolecules from diffusion-based structure prediction models, which typically output highly concentrated distributions around a single static structure.
核心创新
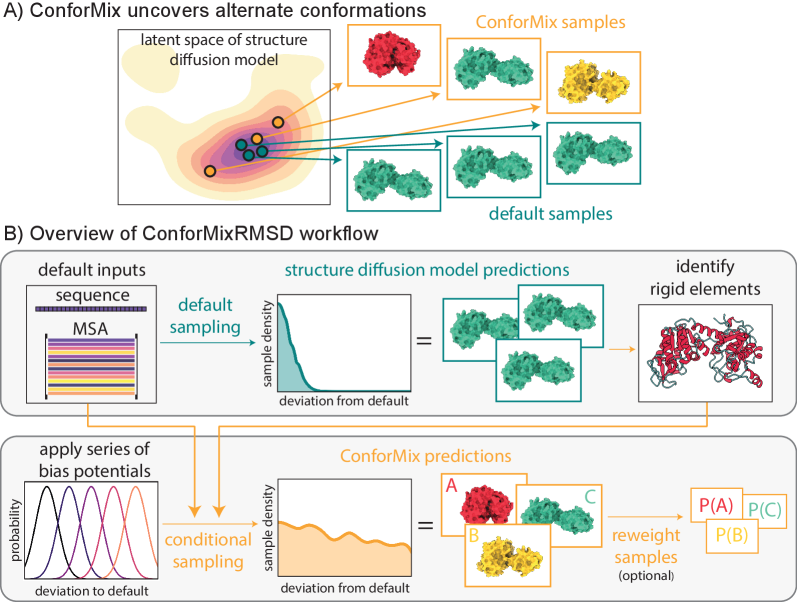
- Methodology Introduces ConforMix, a novel inference-time algorithm combining twisted sequential Monte Carlo (SMC) with automated exploration of the diffusion landscape, enabling asymptotically exact sampling of conditional distributions without additional model training.
- Methodology Presents ConforMixRMSD, an instantiation for automated exploration that biases sampling away from the default prediction using RMSD-based potentials on rigid secondary structure elements, recovering diverse conformations without prior knowledge of degrees of freedom.
- Methodology Applies the multistate Bennett acceptance ratio (MBAR) free energy estimation algorithm to diffusion models for the first time, enabling reconstruction of the unbiased model landscape from conditional samples.
主要结论
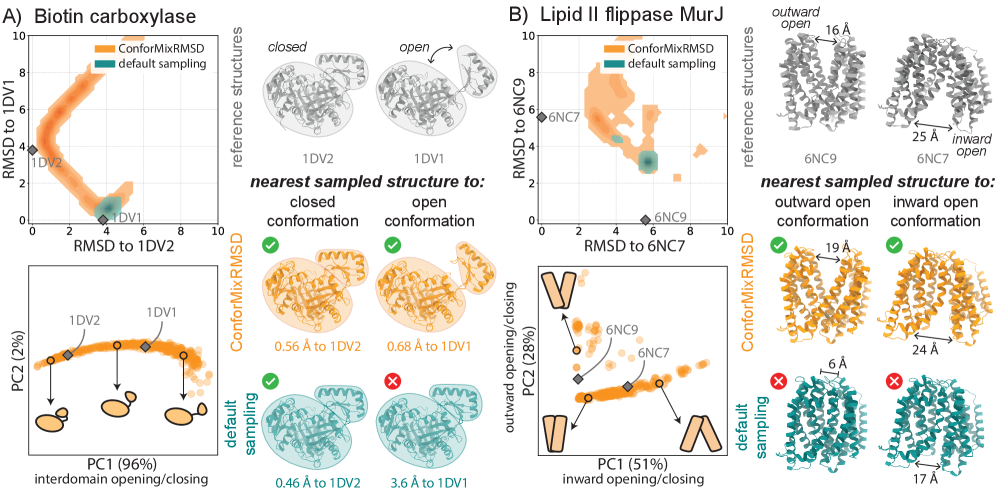
- ConforMixRMSD applied to Boltz-1 (an AlphaFold 3-like model) significantly outperforms MSA-modification baselines (AFCluster, AFSample2, CF-random) in recovering experimentally observed alternative conformations for domain motion (coverage: 0.69 ± 0.15 vs. 0.51 ± 0.17 for best baseline), membrane transporter (0.33 ± 0.23 vs. 0.20 ± 0.20), and cryptic pocket (0.45 ± 0.18 vs. 0.39 ± 0.16) protein sets, as measured by coverage at 50% of reference-to-reference RMSD.
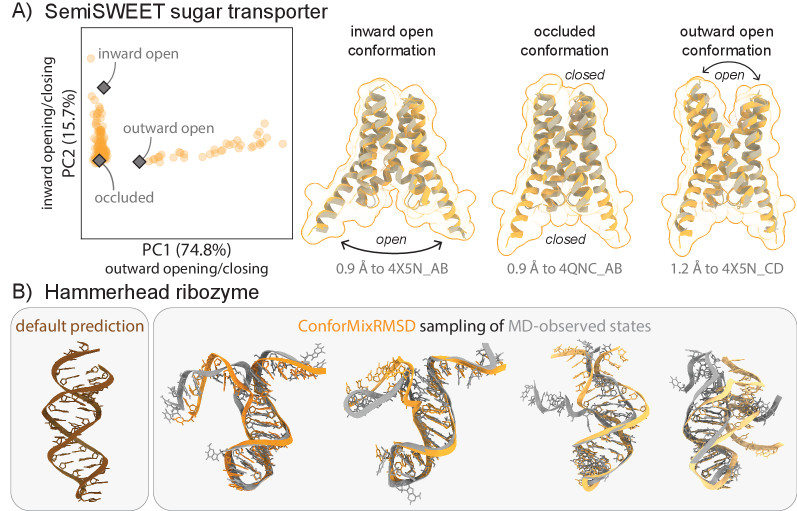
- The method captures biologically relevant conformational transitions (domain motion, transporter cycling, cryptic pocket flexibility) while avoiding unphysical states through filtering based on pLDDT values and clash detection, demonstrating its utility for exploring continuous transitions.
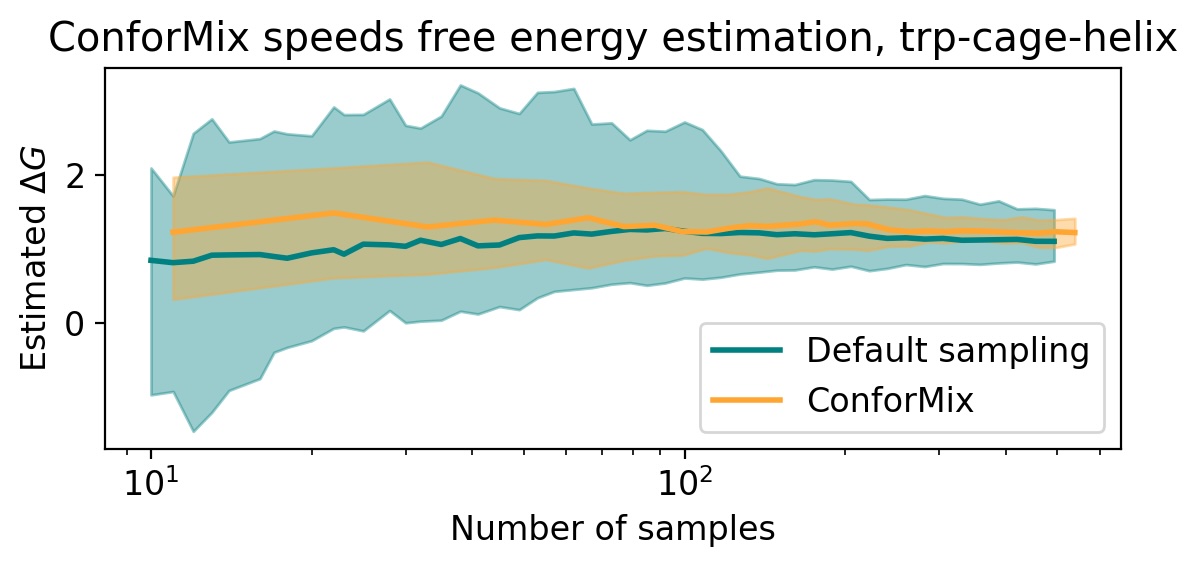
- ConforMix enables efficient free energy estimation when applied to models like BioEmu, boosting the speed of such calculations, and its framework is orthogonal to model pretraining improvements, meaning it would benefit even a hypothetical model that perfectly reproduces the Boltzmann distribution.
摘要: The function of biomolecules such as proteins depends on their ability to interconvert between a wide range of structures or “conformations.” Researchers have endeavored for decades to develop computational methods to predict the distribution of conformations, which is far harder to determine experimentally than a static folded structure. We present ConforMix, an inference-time algorithm that enhances sampling of conformational distributions using a combination of classifier guidance, filtering, and free energy estimation. Our approach upgrades diffusion models—whether trained for static structure prediction or conformational generation—to enable more efficient discovery of conformational variability without requiring prior knowledge of major degrees of freedom. ConforMix is orthogonal to improvements in model pretraining and would benefit even a hypothetical model that perfectly reproduced the Boltzmann distribution. Remarkably, when applied to a diffusion model trained for static structure prediction, ConforMix captures structural changes including domain motion, cryptic pocket flexibility, and transporter cycling, while avoiding unphysical states. Case studies of biologically critical proteins demonstrate the scalability, accuracy, and utility of this method.