
Paper List
-
Ill-Conditioning in Dictionary-Based Dynamic-Equation Learning: A Systems Biology Case Study
This paper addresses the critical challenge of numerical ill-conditioning and multicollinearity in library-based sparse regression methods (e.g., SIND...
-
Hybrid eTFCE–GRF: Exact Cluster-Size Retrieval with Analytical pp-Values for Voxel-Based Morphometry
This paper addresses the computational bottleneck in voxel-based neuroimaging analysis by providing a method that delivers exact cluster-size retrieva...
-
abx_amr_simulator: A simulation environment for antibiotic prescribing policy optimization under antimicrobial resistance
This paper addresses the critical challenge of quantitatively evaluating antibiotic prescribing policies under realistic uncertainty and partial obser...
-
PesTwin: a biology-informed Digital Twin for enabling precision farming
This paper addresses the critical bottleneck in precision agriculture: the inability to accurately forecast pest outbreaks in real-time, leading to su...
-
Equivariant Asynchronous Diffusion: An Adaptive Denoising Schedule for Accelerated Molecular Conformation Generation
This paper addresses the core challenge of generating physically plausible 3D molecular structures by bridging the gap between autoregressive methods ...
-
Omics Data Discovery Agents
This paper addresses the core challenge of making published omics data computationally reusable by automating the extraction, quantification, and inte...
-
Single-cell directional sensing at ultra-low chemoattractant concentrations from extreme first-passage events
This work addresses the core challenge of how a cell can rapidly and accurately determine the direction of a chemoattractant source when the signal is...
-
SDSR: A Spectral Divide-and-Conquer Approach for Species Tree Reconstruction
This paper addresses the computational bottleneck in reconstructing species trees from thousands of species and multiple genes by introducing a scalab...
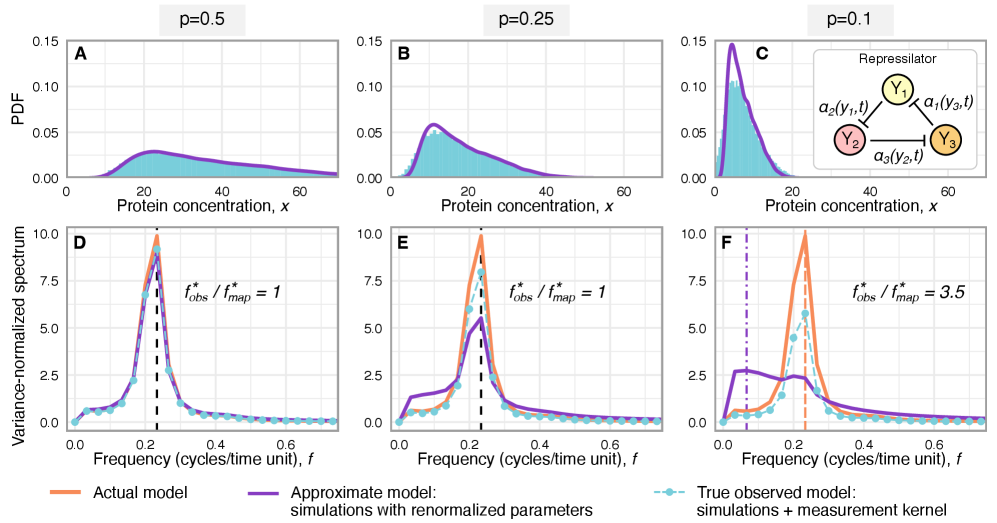
Imperfect molecular detection renormalizes apparent kinetic rates in stochastic gene regulatory networks
Department of Mathematical Analysis and Numerical Mathematics, Comenius University, Slovakia | University of Edinburgh, UK
30秒速读
IN SHORT: This paper addresses the core challenge of distinguishing genuine stochastic dynamics of gene regulatory networks from artifacts introduced by imperfect molecular detection in single-cell experiments.
核心创新
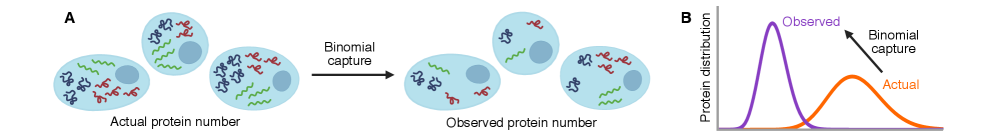
- Methodology Extends the binomial capture model from simple gene expression to general gene regulatory networks (GRNs) with explicit regulation, enabling analysis of technical noise in complex systems.
- Theory Establishes precise mathematical conditions under which technical noise leads to a renormalization (rescaling) of kinetic rates versus when it introduces non-absorbable distortions.
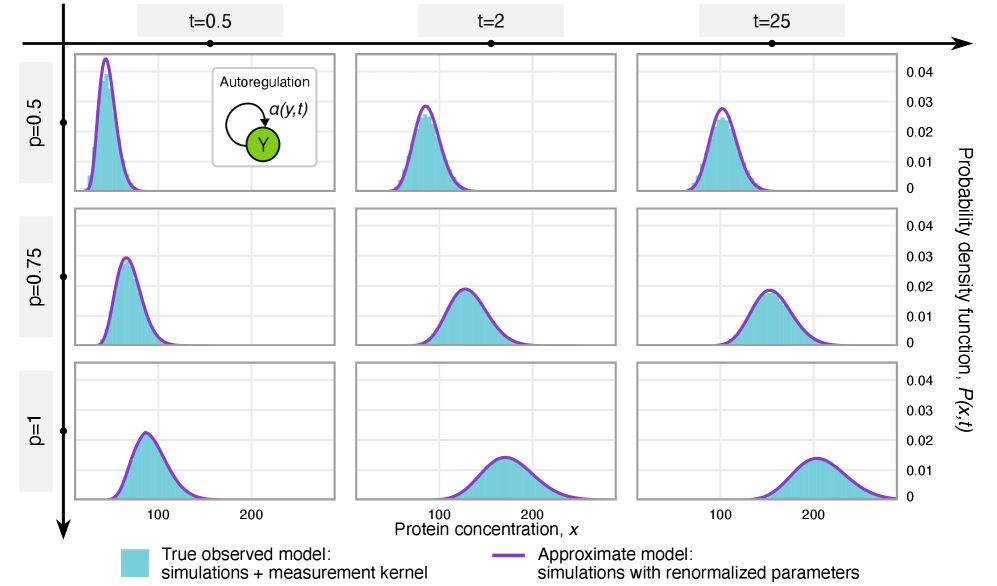
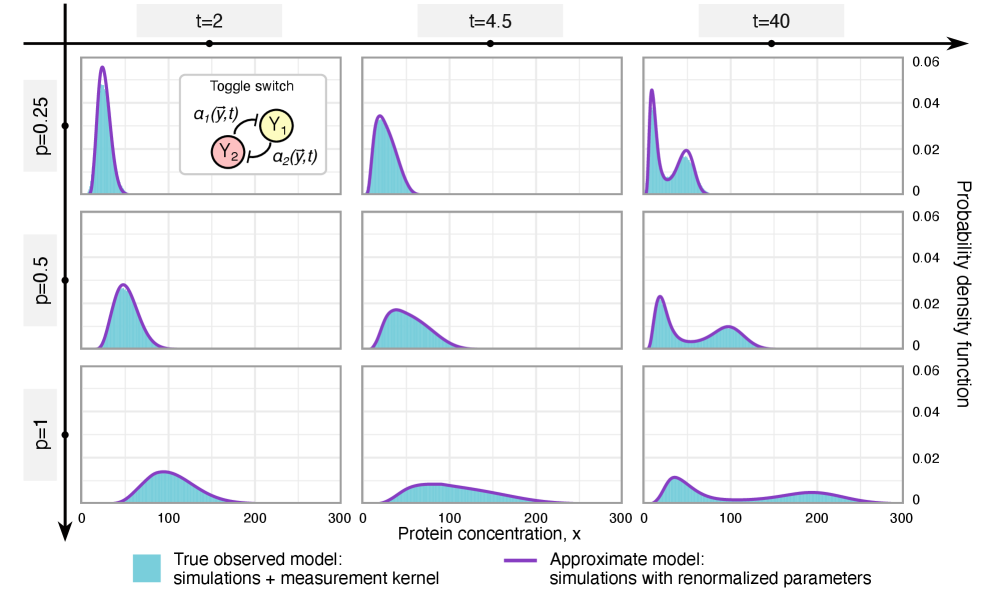
- Methodology Derives results valid for networks of arbitrary connectivity and under time-dependent kinetic rates, significantly generalizing previous steady-state analyses.
主要结论
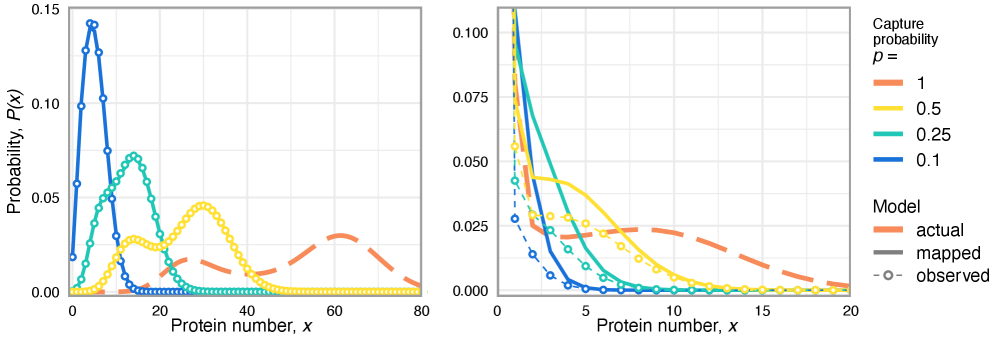
- Technical noise systematically reduces the apparent mean burst size of gene products by a factor of p (the capture probability), e.g., from b(t) to b(t)*p.
- Rate renormalization occurs when promoter-state transitions are on a distinct timescale (much slower/faster) than other reactions or under high transcription factor abundance.
- The framework shows that for the telegraph model, the observed mRNA dynamics are equivalent to the true system with a renormalized transcription rate: k₃(t) → p*k₃(t).
摘要: Imperfect molecular detection in single-cell experiments introduces technical noise that obscures the true stochastic dynamics of gene regulatory networks. While binomial models of molecular capture provide a principled description of imperfect detection, they have so far been analyzed only for simple gene-expression models that do not explicitly account for regulation. Here, we extend binomial models of capture to general gene regulatory networks to understand how imperfect capture reshapes the observed time-dependent statistics of molecular counts. Our results reveal when capture effects correspond to a renormalization of a subset of the kinetic rates and when they cannot be absorbed into effective rates, providing a systematic basis for interpreting noisy single-cell measurements. In particular, we show that rate renormalization emerges either under significant transcription factor abundance or when promoter-state transitions occur on a distinct (much slower or faster) timescale than other reactions. In these cases, technical noise causes the apparent mean burst size of synthesized gene products to appear reduced while transcription factor binding reactions appear faster. These effects hold for gene regulatory networks of arbitrary connectivity and remain valid under time-dependent kinetic rates.